Breast cancer is the most common malignancy in women. Since the seminal work of Perou, Sorlie, and colleagues1,2 in the early 2000s, breast cancer has been stratified into distinct clinically relevant subtypes: luminal cancers that are dependent on hormone receptors (estrogen receptor and progesterone receptor), HER2-enriched tumors harboring HER2 amplification, and basal like tumors which are negative for hormone receptors and HER2. This stratification dictates differential clinical outcomes and choice of therapies. Among these subtypes, estrogen receptor (ER)+ tumors, also referred as luminal breast cancers, are the most common, representing 70% of breast cancers. Breast cancer was first demonstrated to be a hormone-responsive disease by George Beatson in 1896, long before the discovery of ER by Elwood Jensen and Jack Gorski in 1967. These findings ignited the development of endocrine therapies, ushering in the era of personalized medicine. Currently, the ER signaling pathway is targeted clinically mainly with ER antagonist tamoxifen, the ER degrader fulvestrant, or the aromatase inhibitors letrozole and exemestane. Endocrine therapies have significantly improved survival of ER+ breast cancers for decades. However, emergence of resistance to these agents eventually develops in all patients with metastatic disease, and resistance is the primary limiting factor in achieving cures3. Mechanisms of resistance to endocrine therapies, apart from recurrent mutations of ESR1 (the gene coding for ER), are largely unknown.
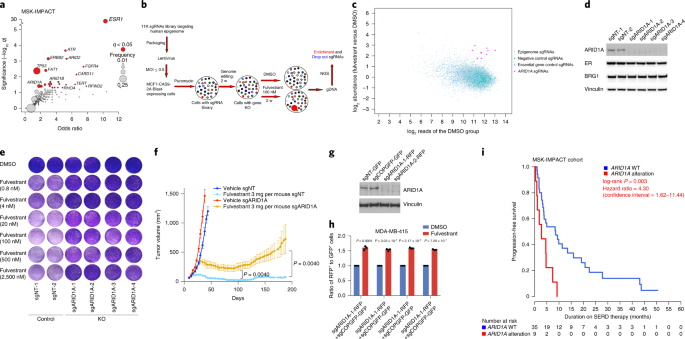
Our goals are to identify the epigenomic drivers of breast tumorigenesis and therapeutic response woven together with the use of computational methods to study gene regulation. In order to identify the genes that may mediate resistance to endocrine therapies, we have analyzed the genomes of over 2,000 breast tumors4. Notably, ARID1A loss of function mutations were enriched in metastatic tumors that have progressed on hormonal therapy. This large institutional clinico-genomic effort unmasked a previously unexpected contribution of chromatin regulatory factors to therapeutic response in breast cancer. Given our long-standing curiosity in how epigenetic modifiers regulate transcription in cancer, the study of ARID1A’s role in breast cancer and therapy response quickly became a strong area of focus. Meanwhile in an initially separate study, a postdoctoral fellow in our laboratory was performing CRISPR/CAS9 genetic screens to identify novel mechanisms of fulvestrant resistance. To our surprise, he also identified ARID1A as the top candidate whose loss determines resistance to the ER degrader fulvestrant. This set of independent observations in patients’ tumors and in CRISPR screens prompted us to join forces to explore the mechanisms whereby ARID1A loss mediates endocrine resistance.
We first confirmed that ARID1A loss mediates fulvestrant resistance using multiple models of ER+ breast cancer including patients tumors harboring ARID1A alterations. We utilized our distinct expertise in computational analyses and epigenetic regulation in breast cancer to generate, analyze, and integrate genome-wide chromatin accessibility, transcriptomic, and chromatin binding assays to examine the global epigenetic consequences of ARID1A loss. We also reached out to a collaborator at Dana-Farber who provided expert advice and performed SWI/SNF chromatin binding assays. These analyses showed that ARID1A loss led to reprograming of the chromatin landscape of breast cancer to regulate the accessibility at luminal lineage determining transcription factors target sites including ER, FOXA1 and GATA3. Interestingly, when we looked at the transcriptomic profile in cell lines and patient tumors with ARID1A loss of function, we found that the signatures looked more like that of basal rather than luminal tumors. Loss of ARID1A-dependent SWI/SNF complex targeting to genomic sites of ER-FOXA1-GATA3 enabled an increased cellular plasticity of luminal cells upon ARID1A inactivation. Thus, ARID1A emerged as a key luminal-lineage regulator whose loss of expression alters luminal differentiation properties and lineage fidelity, leading to loss of dependency on ER and target independence (Figure 1).

Figure 1: Proposed model of ARID1A mediating endocrine therapy sensitivity in ER+ breast cancer
Altogether, our findings may provide an explanation for the clinical observation in which ER+ breast cancer exposed to therapy may eventually switch lineages and become endocrine therapy resistant. To this end, therapeutic pressure may enable the emergence of cells with acquired ARID1A mutations that confer independence from ER therapy. We anticipate that further epigenomic and transcriptomic studies would shed light on therapeutic resistance mediated by lineage switching in cancer and suggest new treatment modalities for these tumors.
In the process of identifying a previously unknown mechanism of resistance to endocrine therapies, we were ultimately reminded of the power of collaboration. By marrying the diverse expertise of collaborators from MSK and Dana-Farber, we hoped to further support advances in personalized medicine by understanding cancer’s biological building blocks.
Our Paper: https://www.nature.com/articles/s41588-019-0554-0.pdf
By Eneda Toska and Christina Leslie
References:
1. Perou, C.M.et al.Molecular portraits of human breast tumours. Nature 406, 747-52 (2000).
2. Sorlie, T.et al.Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A98, 10869-74 (2001).
3. Vasan, N., Baselga, J. & Hyman, D.M. A view on drug resistance in cancer. Nature 575, 299-309 (2019).
4. Razavi, P.et al.The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell34, 427-438 e6 (2018).



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in