
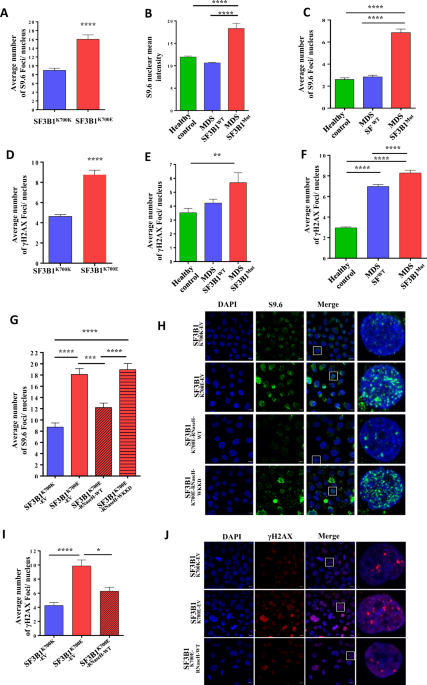
The myelodysplastic syndromes (MDS) are among the most common myeloid malignancies. Pre-mRNA splicing is a fundamental cellular process and it has been shown that genes involved in pre-mRNA splicing (including SF3B1, SRSF2, U2AF1 and ZRSR2) are commonly mutated in MDS and in other myeloid malignancies.1,2 Large-scale RNA sequencing studies have shown that the splicing alterations induced by each mutated splicing factor in MDS largely affect different transcripts, raising the possibility that these mutations may act through other mechanisms in addition to splicing perturbation.3,4 We were intrigued to see in previous studies that mutations in the splicing factors SRSF2 and U2AF1 lead to increased formation of R-loops (RNA-DNA hybrids with a displaced single stranded DNA) in leukemia cell lines, resulting in increased DNA damage, replication stress and activation of the ATR-Chk1 pathway.5,6 In addition, we had recently observed that aberrant splicing of several genes involved in the suppression/regulation of R-loop formation occurs in CD34+ cells of splicing factor mutant MDS patients.3 Given that SF3B1 is the most frequently mutated splicing factor gene in MDS (25-30% of cases), we decided to investigate whether SF3B1 mutations result in elevated R-loop formation in MDS and leukemia cells.
Indeed, we found that increased R-loops and DNA damage occur in association with the presence of SF3B1 mutations in the myeloid leukemia cell line K562, in MDS-patient derived induced pluripotent stem cells and in MDS patient bone marrow CD34+ cells. We then showed activation of the ATR pathway, a major player in the DNA damage response, in SF3B1 mutant hematopoietic cells, raising the exciting prospect that this could represent a therapeutic vulnerability. Our next experiments supported this hypothesis, as we found that SF3B1 mutant K562 cells and primary MDS patient bone marrow cells showed preferential sensitivity to the ATR inhibitor VE-821 and to UCN-01, an inhibitor of Chk1 (a critical substrate of ATR), suggesting that ATR-Chk1 activation is important for the survival of SF3B1 mutant cells. Notably, a phase 1 clinical trial to assess the safety and efficacy of another ATR inhibitor (AZD6738) in patients with MDS and chronic myelomonocytic leukemia is currently underway in the US (NCT03770429).
Several groups have shown that splicing modulators have some efficacy in splicing factor mutant leukemias in pre-clinical studies.7 We therefore chose to treat SF3B1 mutant cells with the splicing modulator Sudemycin D6 in combination with an ATR or Chk1 inhibitor. Importantly, we found that the effects of VE-821 or UCN-01 on SF3B1 mutant K562 cells and primary MDS patient bone marrow cells were enhanced by Sudemycin D6, indicative of synergy between these drugs. This finding has translational implications and warrants further pre-clinical studies.
Our study on mutant SF3B1 together with previous studies by others on mutant U2AF1 and SRSF2 demonstrate that different mutated splicing factors have convergent effects on R-loop elevation leading to DNA damage in hematopoietic cells. It is currently not know why splicing factor mutant cells have a clonal advantage in human bone marrow, and this R-loop induced DNA damage may be a contributing factor.
Our data show for the first time that Chk1 inhibition, alone or in combination with splicing modulators, may represent a novel therapeutic strategy to target SF3B1 mutant cells in hematological malignancies. It would be interesting to determine whether this approach could be extended to therapeutically target other types of cancers known to harbor SF3B1 mutations.
Written by Andrea Pellagatti and Jacqueline Boultwood
References
1. Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478:64-9.
2. Papaemmanuil E, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011; 365:1384-95.
3. Pellagatti A, et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood 2018; 132:1225-40.
4. Shiozawa Y, et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat Commun 2018; 9:3649.
5. Chen L, et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol Cell 2018; 69:412-25 e6.
6. Nguyen HD, et al. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res 2018; 78:5363-74.
7. Obeng EA, et al. Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer Discov 2019; 9:1493-1510.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in