In endocrine cells, the cAMP-PKA signaling pathway is pivotal for the regulation of the synthesis and secretion of hormones that control stress and growth response. Therefore, it is not surprising that activated PKA is a hallmark of endocrine tumors including adrenal and GH-secreting pituitary tumors. It is intriguing that despite several sequencing efforts, genomic alterations that could lead to PKA activation such as activating GNAS1 (aka gsp) mutations are found only in a subset of cases.
Several years ago, while working on the role of angiogenic factors in pituitary tumor pathophysiology, we were puzzled about the lower vascular density in the vast majority of pituitary tumors compared to healthy pituitary gland. When presenting work on this topic at conferences or even in internal lab meetings the same questions would inevitably come up: why do these tumors have fewer vessels than normal pituitary tissue? Isn’t angiogenesis a characteristic of tumors meaning they should have more vessels? And what kind of advantage does this reduced vascularization give to the tumor? As time progressed, these questions remained largely philosophical.
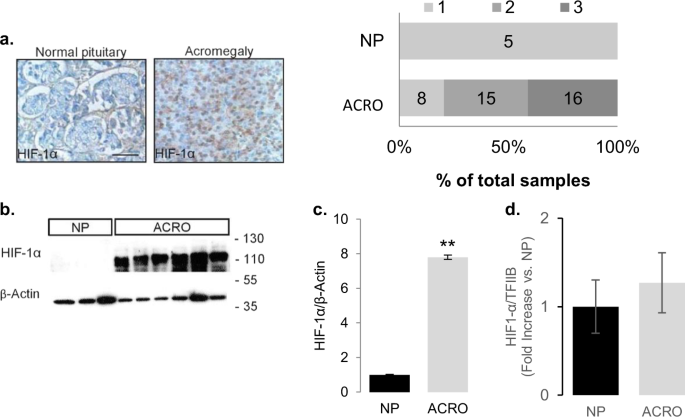
Several years later while working on the crosstalk of nutrient sensors and the cAMP-PKA cascade in somatotroph cells, we came across a remarkable review of Gregg Semenza on metabolic responses to tumor hypoxia and HIF-1α. This made us wonder whether what we are witnessing in our tumors is relative tissue hypoxia, so we promptly looked at HIF-1α expression and realized that GH-secreting pituitary tumors have the lowest vascular density and the highest HIF-1α protein levels. The next -inevitable- question was whether the hypoxic state and increased HIF-1a may be responsible for the PKA overactivation characteristic of these endocrine tumors. Indeed, we found that hypoxia and HIF-1a trigger GH synthesis from somatotroph tumor cells and that PKA activation was involved in this process. We then started dissecting the signaling cascade of PKA using GH as a readout and we were nearly at the end of the pathway, not yet having found a clear target of HIF-1α when we examined the constitution of the PKA holoenzyme and identified the regulatory subunit PRKARIIB as the likely culprit. From here, additional experiments were performed to test the robustness of this finding in the context of GH synthesis under hypoxic conditions, which ultimately confirmed the role of the PKA subunit in promoting hypoxia-induced GH synthesis.
Looking back at the course of this project, which indeed included several stumbling stones, one thing seemed to have provided the impetus which put us on the right track: first take a step back and understand the basic physiology of the cell you are studying. This may seem over-simplistic, however in the process of unraveling an unusual intersection of two seemingly unrelated pathways, this question always brought us back on track.
Co-written by Kristin Lucia and Marily Theodoropoulou

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in