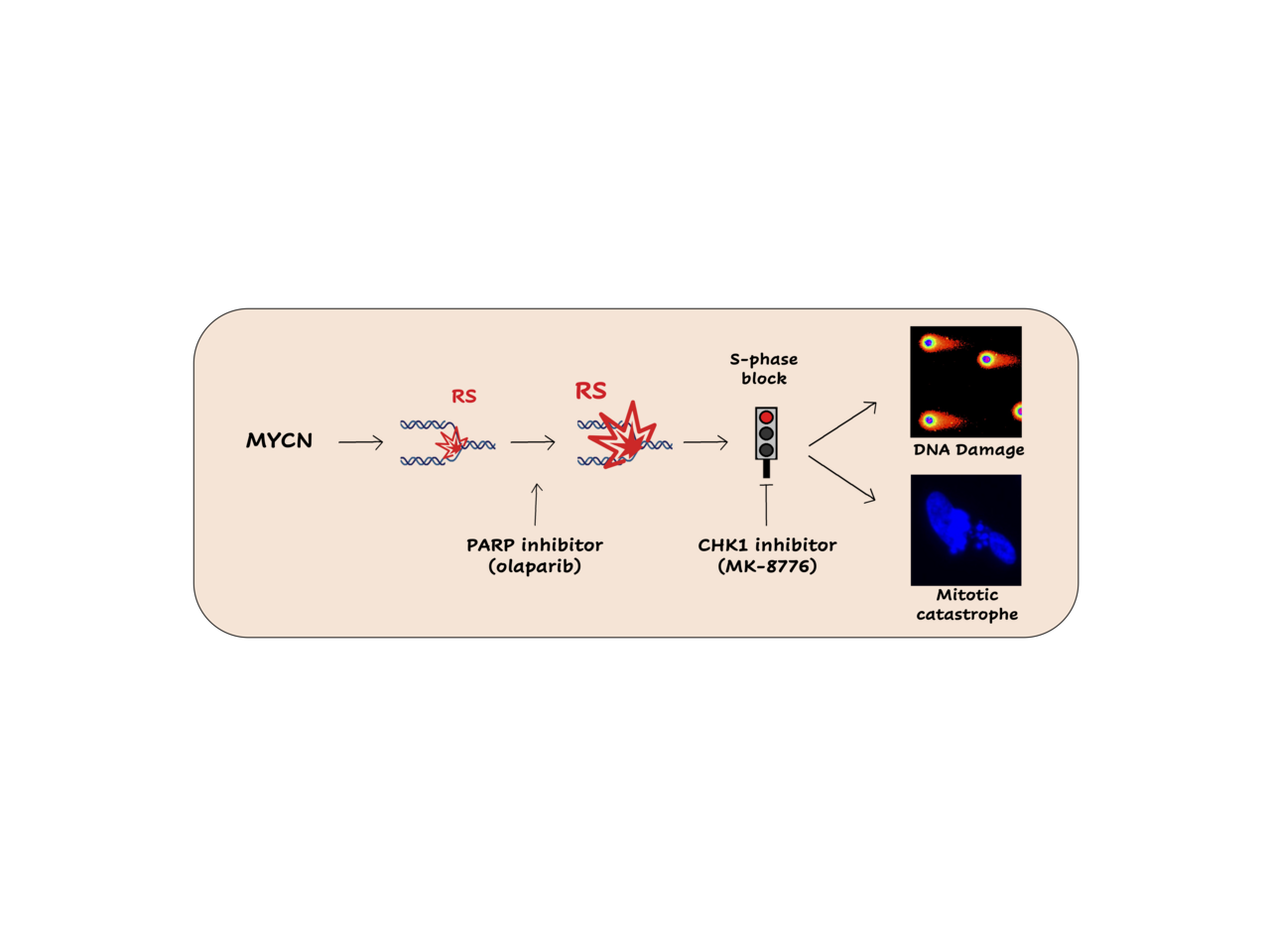

MYCN-dependent tumors mostly occur as aggressive and deadly neoplasias and are very poorly responsive to current therapies. The identification of alternative vulnerabilities of these tumors has been one of the main fields of interest of Giannini’s lab over the last 10 years, during which targeting MYCN-dependent replication stress (RS) response is emerging as a potentially useful strategy. Already in 2017 we demonstrated that PARP inhibition increases MYCN-dependent RS and leads MYCN-amplified neuroblastoma cells to mitotic catastrophe, despite the activation of a cell cycle checkpoint driven by CHK1 in S-phase (1). Furthermore, we showed that CHK1 inhibition combined with PARP inhibition increases cell death (1). Therefore, we sought to explore the therapeutic potential of these observations with the aim to find out a new and effective therapy for MYCN-amplified tumors.
In our recently published study in Oncogene, we determined the biochemical and biological consequences of CHK1i/PARPi combination in MYCN-dependent tumors, in vitro and in vivo preclinical models.
It is well-established that, due to the oncogene-dependent RS, Myc/MYCN-dependent tumors are strictly addicted to the ATR-CHK1 pathway (2). Coherently, we reported a higher CHK1 expression in human MYCN-amplified (MNA) neuroblastoma cell lines and primary tumor samples compared to non-MNA conditions, as well as in SHH-type medulloblastomas compared to normal cerebella, in preclinical animal models and human patients. Importantly, MNA and MYCN overexpressing (MYCN-OE) cell lines showed higher sensitivity to CHK1 inhibition.
All together our data supported the potential effectiveness of CHK1i/PARPi combination in the treatment of MYCN-dependent tumors.
It is emerging that CHK1 inhibitors alone or in combination with chemotherapy are associated with toxicity in several phase 1/2 clinical trials (3,4), strongly limiting their development and employment. To limit the toxicity related to CHK1 inhibitors, in our work we used suboptimal doses of the CHK1 inhibitor MK-8776 showing it efficiently impairs the S-phase checkpoint and the DNA repair by homologous recombination (HR) triggered by the PARP inhibitor Olaparib, leading to the accumulation of RS-born DNA damage, DDR and cell death, in MNA and MYCN-OE cells. Thus, we decided to test the efficacy of MK-8776/Olaparib combination in vivo. Remarkably, we observed that suboptimal doses of MK-8776 plus Olaparib most efficiently antagonized the growth of different MNA neuroblastoma preclinical models in vivo, including subcutaneous and orthotopic xenografts, where the CHK1i/PARPi combination increased the rate of apoptosis and reduced tumor vascularization. Similarly, the combination of low doses of MK-8776 plus Olaparib also efficiently inhibited the growth of SHH-driven medulloblastoma in vitro and in vivo, confirming its efficacy in a different MYCN-driven tumor model.
Importantly, we did not detect significant organ-specific or systemic toxicities, which are often seen in pharmacological associations of CHK1 inhibitors with DNA damaging drugs, indicating that pharmacological combinations with PARP inhibitors might improve the therapeutic index of CHK1 inhibitors by reducing dose-related side effects.
Interestingly, we noticed that the IMR-32 cell line, carrying the putatively pathogenic p.Val2716Ala ATM mutation, was the most sensitive line to the MK-8776/Olaparib combination. Since it is well-established that PARP inhibitors are strongly effective in the absence of important genes relevant for homologous recombination (such ATM) (5, 6), we evaluated whether this specific ATM mutation contributed to enhance the sensitivity to the CHK1i/PARPi combination in this cell line. Interestingly, the genetic correction of the ATM mutation by CRISPR/Cas9-based technology did not reduce the effectiveness of the treatment, highlighting that the high efficacy of CHK1i/PARPi combination was not related to ATM status.
To conclude, we report that a chemo-free approach combining CHK1 and PARP inhibitors antagonizes the growth of MYCN-driven tumors in vitro and in preclinical models in vivo without major toxicities. Considering the advanced state of development and testing of the drugs, this strategy could be promptly translated into clinical trials.
Figure 1. MK-8776/olaparib combination increases MYCN-dependent replication stress leading to cell death by mitotic catastrophe in vitro and strongly reducing tumor growth in vivo

References
1) Colicchia V, Petroni M, Guarguaglini G, Sardina F, Sahun-Roncero M, Carbonari M, et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene. 2017;36:4682–91.
2) Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Mycdriven tumors. Nat Struct Mol Biol (Res Support, Non-U S Gov’t). 2011;18:1331–5.
3) Cash T, Fox E, Liu X, Minard CG, Reid JM, Scheck AC et al. A phase 1 study of prexasertib (LY2606368), a CHK1/2 inhibitor, in pediatric patients with recurrent or refractory solid tumors, including CNS tumors: a report from the Children’s Oncology Group Pediatric Early Phase Clinical Trials Network (ADVL1515). Pediatric Blood Cancer. 2021;68
4) Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol. 2015;33:1060–6.
5) Sanmartin E, Munoz L, Piqueras M, Sirerol JA, Berlanga P, Canete A, et al. Deletion
of 11q in Neuroblastomas Drives Sensitivity to PARP Inhibition. Clin Cancer Res. 2017;23:6875–87.
6) Takagi M, Yoshida M, Nemoto Y, Tamaichi H, Tsuchida R, Seki M, et al. Loss of DNA Damage Response in Neuroblastoma and Utility of a PARP Inhibitor. J Natl Cancer Inst. 2017;109:11



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in