Multiple myeloma progression involves the spread and seeding of cancerous cells to different body sites. At relapse, the multiple myeloma clonal composition at different sites varies, i.e. the genetic aberrations harbored by tumor cells differ. However, it is not known whether this reflects the expansion of previously undetected pre-existing cancer or spread from other sites. Furthermore, the genetic evolution over an individual’s lifetime at different sites of disease and how therapy contributes to this change has not been investigated in a systematic manner.
To address this, we collected 25 samples at autopsy from 4 patients with relapsed multiple myeloma within the Last Wish Program at Memorial Sloan Kettering Cancer Center. The Last Wish Program is an ongoing research biospecimen protocol which permits the postmortem collection of tissue and other samples from deceased patients, from whom consent for the protocol was obtained antemortem. To capture the myeloma complexity and heterogeneity we performed whole genome sequencing on all these samples. Doing so, we demonstrated that each metastatic site within the same individual had a distinct trajectory of evolution, punctuated by simple and complex genetic abnormalities accumulated over time. By examining the landscape of mutational processes (i.e. mutational signatures) active in each individual site, we observed that chemotherapeutic drug melphalan and platinum significantly contribute to the global mutational burden. In contrast to newly diagnosed patients, (never exposed to melphalan and platinum), we identified that chemotherapy-induced mutations account for 25% of all nonsynonymous mutations in relapsed multiple myeloma.
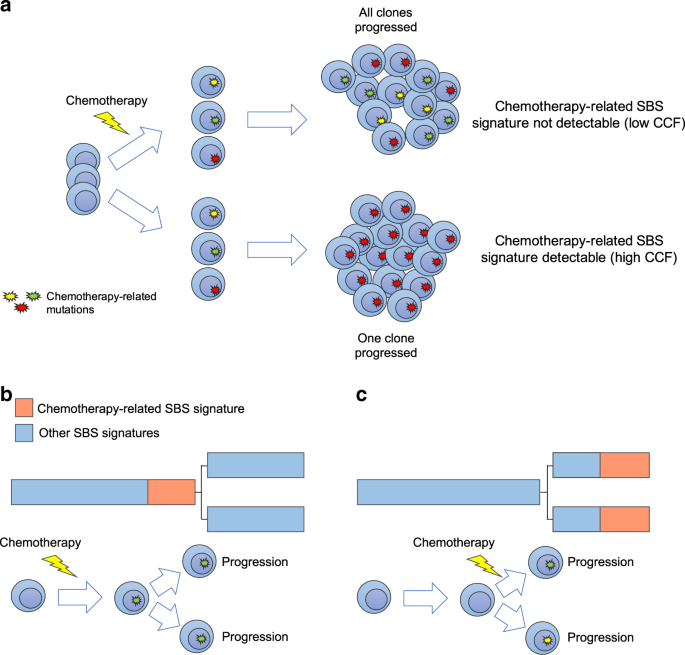
Exposure to distinct chemotherapies is known to introduce hundreds of mutations in each surviving cancer cell, with the exact mutational catalog being different in each cell (Figure 1). Therefore, mutations caused by chemotherapy may be detected only if a single cell containing these mutations repeatedly divides to form the majority of cancer cells at a particular metastatic site (the dominant clone) (Rustad et al. Nat Comm 2020). The mutational signature linked to chemotherapy thus represents a unique single-cell barcode linked to a known time point in each patient’s life.
We used this concept to show that multiple myeloma spread around the body at clinical relapse is often driven by the survival and subsequent expansion of a single myeloma cell following treatment with high dose melphalan therapy and autologous stem cell transplant. In fact, in three out of four cases all disease localization shared the same melphalan-induced mutational catalog, suggesting a common origin from the same surviving single cell.
Looking at the clinical and radiological history of these patients, we observe how some of these multi-site localizations occurred in a short time frame (e.g. <2 years). This pattern of timing suggests that seeding of a single clonal cell around the body can occur rapidly at relapse. To further investigate this hypothesis, and to explore potential differences between pre- and post-treatment disease seeding, we combined our cohort with a previously published cohort of newly diagnosed and relapsed multiple myeloma whole exomes (n=125 from 51 patients; Rasche et al. Nat Comm 2017). Comparing the contribution of mutational signatures associated with aging (SBS1, described as clock-like; Alexandrov et al. Nat Gen 2015) between the branches and the trunk of the phylogenic tree of each patient, we observed that the ratios of SBS1 between branches and the trunk were significantly higher in the treatment-naïve patients compared to those observed in the relapsed WXS and WGS cases, consistent with a subclonal diversification having occurred over a long period of time. These findings support the model in which multiple myeloma seeding can be accelerated following high-dose treatment in comparison to that seen during spontaneous evolution.
These data highlight the importance of considering the complex spatial and temporal heterogeneity of multiple myeloma in the evaluation of treatment-response and in minimal residual disease assessment and provide a strong rationale for comprehensive characterization of the multiple myeloma genomic complexity to enhance clinical decision making.
Figure 1. The development of chemotherapy-related mutational signatures. a) A schema summarizing the single cell expansion model. In this model, chemotherapy-related mutational signatures will be detectable only if one cancer cell is selected and takes the clonal dominance. (SBS = single base substitution; CCF = cancer-cell fraction). b-c) Two possible scenarios for the development of chemotherapy-related mutational signatures in two different disease localizations. In (b), chemotherapy is delivered to the trunk of the phylogenetic tree prior to any branching, while in (c) chemotherapy is delivered after branching. The phylogenetic tree trunk and branch lengths represent the mutational load.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in