

Metabolic reprogramming is one of the most important hallmarks of cancer (Hanahan and Weinberg, 2011). Tumor cells undergo a series of metabolic reprogramming to meet the demands for rapid proliferation and growth. The Warburg Effect proposed by Otto Warburg in the 1920s points out that even under aerobic conditions, tumor cells prefer anaerobic glycolysis to produce energy and biosynthetic building blocks for cell proliferation (Warburg, 1956). Since then, tremendous research efforts have been made to understand the underlying molecular mechanisms of metabolic reprogramming and its clinical significance. Recent studies have unveiled a diverse mechanisms for cancer cells to achieve metabolic reprogramming, such as gene mutations, epigenetic alterations, protein-protein interactions, and the interaction between metabolic enzymes and metabolites (Bose et al., 2020; Hopkins et al., 2020; Wang et al., 2020; Zhu and Thompson, 2019).
Aldolases catalyze the cleavage of fructose-1,6-bis-phosphate (FBP) to generate dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (GAP) in glycolysis, as well as the reverse reaction of DHAP and GAP to FBP in gluconeogenesis. Recent years have witnessed increasing research interests on Aldolases in glucose metabolism in cancer and other human diseases (Chang et al., 2018). For example, Dr. Wulf’s group at Harvard University found that PI3K regulates glycolysis though mobilization of aldolase from the actin cytoskeleton (Hu et al., 2016). A recent study from Dr. Sheng-Cai Lin’s laboratory at Xiamen University, China, found that FBP’s binding to aldolase mediates glucose sensing by AMPK independent of AMP levels (Zhang et al., 2017). Aldolase B (Aldob) is abundantly expressed in the liver, small intestine, and kidney. Aldob has been extensively studied in fructose metabolism and human mutations lead to hereditary fructose intolerance (HFI) (Oppelt et al., 2015). A recent work from Dr. Xiling Shen’s laboratory at Duke University discovered that hepatic Aldob-mediated fructose metabolism drives colon cancer metastasis to the liver (Bu et al., 2018). Although previous studies found that Aldob is down-regulated in liver cancer tissues and its expression is reversely correlated to clinical prognosis for liver cancer patients, the molecular mechanisms and clinical significance of Aldob downregulation in hepatocellular carcinoma (HCC) remain elusive.
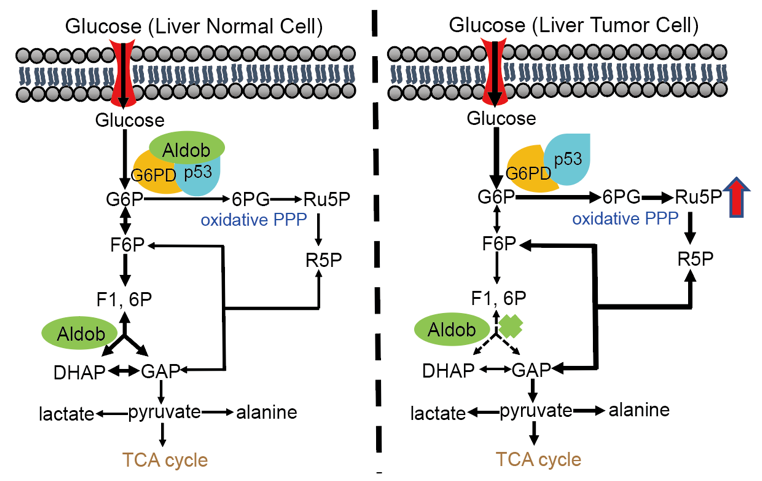
On July 6, 2020, our team published a research paper in Nature Cancer entitled "Aldolase B Suppresses Hepatocellular Carcinogenesis by Inhibiting G6PD and Pentose Phosphate Pathways" (https://www.nature.com/articles/s43018-020-0086-7). This study has discovered a novel mechanism by which Aldob regulates liver cancer metabolic reprogramming. In this study, we started out from gene expression microarray analysis of paired human HCC tissues and found that the expression of Aldob was significantly downregulated, whereas glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of the pentose phosphate pathway (PPP), was significantly upregulated in tumor tissues compared to peripheral normal tissue. Furthermore, a low expression of Aldob and high G6PD expression was positively associated with short overall survival and poor prognosis for HCC patients. A multivariate analysis demonstrated that Aldob is an independent risk factor for HCC. Using N-Nitrosodiethylamine (DEN)-induced HCC mouse model, they further found that global or liver specific knockout (KO) of Aldob promotes hepatocellular carcinogenesis through enhancing G6PD activity to increase the metabolic flux to PPP, while re-expression of Aldob in liver-specific Aldob KO mice suppresses HCC. Consistently, pharmacological inhibition or genetic knocking down G6PD attenuates the tumor formation. All these in vivo data suggest that Aldob opposes HCC through regulation of G6PD and PPP. They further demonstrated that in normal liver cells, Aldob interacts with G6PD to inhibit its activity, and the presence of Aldob enhances the inhibitory effect of tumor suppressor p53 on G6PD through a protein complex containing Aldob, G6PD and p53 to regulate cell metabolism. In cancer cells, however, Aldob downregulation releases G6PD activity and attenuates the inhibitory effect of p53 on G6PD, leading to the increased metabolism of PPP to meet the need for rapid growth of cancer cells.
In summary, our current study has defined a new molecular mechanism for cancer cell metabolic reprogramming: the interaction of metabolic enzymes Aldob and G6PD regulates cell metabolism in glycolysis and PPP, whereas the loss of Aldob in tumor cells leads to rewire metabolic pathways to favor tumor growth. The study found a new non-enzymatic function of Aldob, highlighting a clinical significance in diagnosis and treatment of liver cancer. However, it remains to be investigated what the upstream factors are in regulating the Aldob expression and how Aldob is gradually lost during the progression of HCC.
The graduate students Drs. Min LI and Xuxiao HE from YIN laboratory, and Dr. Weixin GUO from The Eastern Hepatobiliary Surgery Hospital, Shanghai, are the co-first authors for this work. Prof. Huiyong YIN is the lead contact for this article. Dr. Yongzhen TAO, a senior research assistant from YIN Laboratory, and Dr. Shuqun CHENG, a chief physician from The Eastern Hepatobiliary Surgery Hospital are designated as co-corresponding authors. The authors acknowledged the help from Dr. Dangsheng LI and Prof. Jianping Ding from the Center for Excellence in Molecular Cell Science, CAS, Prof. Yu LI from SINH, CAS, and Prof. Shengcai LIN and Shuhai LIN from Xiamen University. This work was funded by the National Natural Science Foundation of China, the Chinese Ministry of Science and Technology, and CAS.

Figure Aldob inhibits liver cancer by directly interacting with G6PD and enhancing p53-mediated inhibition of G6PD
References:
Bose, S., Allen, A.E., and Locasale, J.W. (2020). The Molecular Link from Diet to Cancer Cell Metabolism. Molecular Cell.
Bu, P., Chen, K.Y., Xiang, K., Johnson, C., Crown, S.B., Rakhilin, N., Ai, Y., Wang, L., Xi, R., Astapova, I., et al. (2018). Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab.
Chang, Y.C., Yang, Y.C., Tien, C.P., Yang, C.J., and Hsiao, M. (2018). Roles of Aldolase Family Genes in Human Cancers and Diseases. Trends Endocrinol Metab 29, 549-559.
Hanahan, D., and Weinberg, Robert A. (2011). Hallmarks of Cancer: The Next Generation. Cell 144, 646-674.
Hopkins, B.D., Goncalves, M.D., and Cantley, L.C. (2020). Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nature reviews Endocrinology, 10.1038/s41574-41020-40329-41579.
Hu, H., Juvekar, A., Lyssiotis, C.A., Lien, E.C., Albeck, J.G., Oh, D., Varma, G., Hung, Y.P., Ullas, S., Lauring, J., et al. (2016). Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 164, 433-446.
Oppelt, S.A., Sennott, E.M., and Tolan, D.R. (2015). Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Molecular Genetics and Metabolism 114, 445-450.
Wang, Y.-P., Li, J.-T., Qu, J., Yin, M., and Lei, Q.-Y. (2020). Metabolite sensing and signaling in Cancer. Journal of Biological Chemistry.
Warburg, O. (1956). On the origin of cancer cells. Science 123, 309-314.
Zhang, C.-S., Hawley, S.A., Zong, Y., Li, M., Wang, Z., Gray, A., Ma, T., Cui, J., Feng, J.-W., Zhu, M., et al. (2017). Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112-116.
Zhu, J., and Thompson, C.B. (2019). Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol 20, 436-450.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in