
Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer. This disease is almost always fatal, with a ten-year survival rate of around one percent. So far, significant treatment responses reached only the classical subtype of the disease, characterized by an epithelial morphology and gene expression program. Mesenchymal PDAC, showing an undifferentiated morphology and basal-like transcriptional profile, is the most lethal subtype and is refractory even to the most aggressive treatment options. Immunotherapies, which demonstrated to be highly effective in other tumor entities, do not work in this tumor subtype. On one hand this is probably due to the relatively low number of mutations these tumors harbor, resulting in a low amount of immunogenic neo-antigens; on the other the tumor microenvironment (TME) of mesenchymal PDAC is highly immunosuppressive, preventing the infiltration of T cells into the tumor. For this reason, we refer to these tumors as immunologically “cold”.
Over 90% of PDAC patients show an activating mutation in KRAS. Recently, several reports have shown that an increased gene-dosage (iGD) and expression of mutant KRAS (KRAS-mut) drive the disease, with the mesenchymal basal-like subtype displaying the highest KRAS-mut gene-expression levels (Chan-Seng-Yue, Nature Genetics, 2020 10.1038/s41588-019-0566-9; Mueller, Nature, 2018 10.1038/nature25459). In this study, we set out to develop a combination therapy for mesenchymal PDAC targeting KRAS-driven tumor cell intrinsic signaling and in parallel reprograming the TME.
Initially, we evaluated whether targeting MEK, the canonical downstream effector of KRAS, is effective in mesenchymal PDAC, which shows the highest KRAS-mut iGD and gene expression levels. To this end, we screened a large panel of PDAC cell lines belonging to both classical and mesenchymal subtypes with the clinically approved MEK inhibitor (MEKi) trametinib. Contrary to our hypothesis mainly classical cell lines were sensitive to MEKi, whereas almost all mesenchymal cells showed remarkable resistance.
Based on this observation, we postulated that additional pathways need to be blocked in this highly aggressive PDAC subtype to achieve meaningful therapeutic responses. Therefore, we carried out a high-throughput drug screen combining a total of 418 different compounds and trametinib as anchor. We used human and mouse primary PDAC cell cultures to identify drugs that synergize with trametinib in mesenchymal PDAC. The multikinase inhibitor nintedanib, already clinically approved for the treatment of idiopathic pulmonary fibrosis, was very effective in combination with trametinib in vitro. Importantly, it showed selectivity for mesenchymal PDAC tumor cells. Indeed, we observed that the treatment with both trametinib and nintedanib (T/N) led to cell cycle arrest and cell death in this PDAC subtype, indicating that broad multikinase targeting is essential to achieve therapeutic responses. This was not the case for classical PDAC, for which treatment with T/N was even antagonistic in some cell cultures.
Our in vitro findings brought us to explore the combination treatment in vivo, in syngeneic orthotopic transplantation models of classical and mesenchymal PDAC. Mesenchymal PDAC benefitted from the treatment with the drug combination also in vivo – with strong reduction in tumor volume and doubled survival. Unexpectedly, and contrary to the in vitro setting, we observed a response also in the classical subtype; however, we identified that this effect was mostly trametinib mediated.
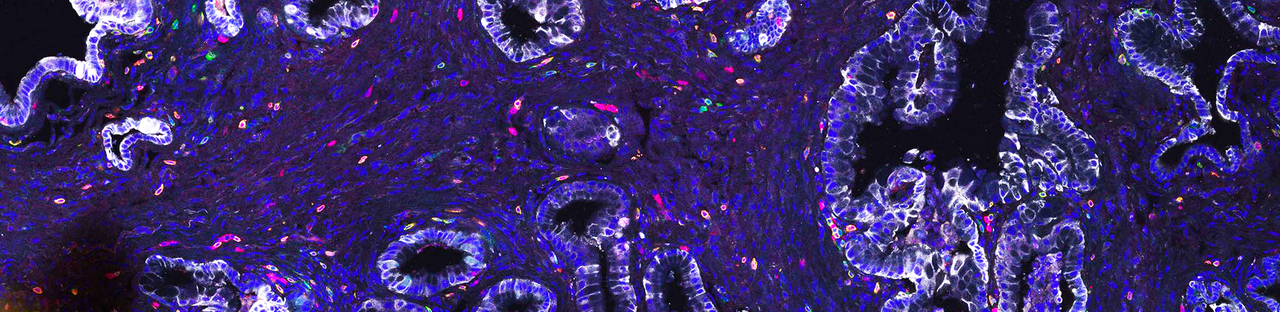
Next, we investigated treatment-induced changes in cell types composing the TME. One of the strongest differences we observed across subtypes and treatment conditions was a remarkable change in T cell composition. Treatment with T/N strongly increased the infiltration of activated CD8+ T cells into mesenchymal tumors. Classical tumors showed treatment induced senescence-associated-secretory-phenotype, recently connected to treatment-induced immune activation. However, in our context they displayed features of immune exclusion – showing only a moderate enrichment of T cells at the tumor margins.
Additional changes were detected in macrophage and neutrophil composition. Notably, in both classical and mesenchymal subtypes, we did not observe a strong change in the number of macrophages, rather in their polarization state: they changed from being mostly pro-tumorigenic M2- to an anti-tumorigenic M1-like state. Moreover, uniquely to classical tumors we observed an increase in neutrophil recruitment upon treatment.
To investigate whether the observed T cell infiltration could sensitize mesenchymal pancreatic carcinomas to immune checkpoint inhibitors, we applied anti PD-L1 immunotherapy. The T/N+anti PD-L1 triple treatment significantly improved the overall response of mesenchymal PDAC leading to a clear survival advantage in comparison to the double treated cohorts.
To systematically analyze therapy-induced TME reprogramming, we performed single cell RNA sequencing of both classical and mesenchymal tumors. First, we analyzed treatment-induced changes in the tumor cells of both subtypes. Gene set enrichment analysis revealed treatment-induced DNA-damage, which has been previously shown to activate immune responses. Further, we could see induction of antigen processing and cross-presentation, accompanied by an exclusive and striking enrichment for interferon signaling signatures in mesenchymal PDAC. Next, we focused on the TME and the T cell compartment. T/N treated tumors presented a strong increase in T cells with functional cytotoxic and effector T cell gene expression signatures. The addition of anti-PD-L1 resulted in a further increase of cytotoxic and effector T cells within these tumors. In contrast, classical tumors displayed a decrease of regulatory T cells.
How are these dynamic changes in the T cell composition regulated? To address this question, we analyzed the secretomes of T/N treated classical and mesenchymal tumor cells. We identified that the combination therapy reprogrammed the immunosuppressive mesenchymal cancer cell secretome and downregulated cytokines and chemokines, including CCL2 and CSF1, capable of attracting and inducing expansion of macrophages and myeloid derived suppressor cells. In parallel, it induced secretion of T cell modulators, such as CXCL16 and CXCL12, important for T cell recruitment.
Taken together, our study shows that reprogramming of the immunologically “cold” tumor stroma of the highly aggressive mesenchymal PDAC subtype is possible. This TME switch can be exploited therapeutically by adding anti PD-L1 immunotherapy. These results are a first, important step, in the targeted treatment of mesenchymal PDAC, for which there are currently no efficient therapeutic options.

Schematic representation summarizing our findings



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in