
A tumor consists of millions of cells, with a wide range of genetic alterations and phenotypical manifestations, a phenomenon denoted as intratumoral heterogeneity. Multiple subpopulations (subclones) of cells with different genetic profiles will be present in the tumor and the prevalence of each of these populations can change over time and space. Cancer therapies or other changes in the tumor microenvironment may result in the death of some cancer cells, while others thrive, akin to Darwinian selection at the cellular level.
One of the most common causes of death in cancer is a tumor that no longer responds to treatment. This is further complicated by widespread metastatic dissemination, making surgery and radiation therapy unfeasible. To save the patients that today die of their disease, new approaches are needed that consider the dynamical evolutionary aspects of tumors. The importance of considering cancer cell evolution in development of novel treatment strategies for cancer, was proposed already 40 years ago by Peter Nowell1. In the last decade, multiple approaches taking evolutionary aspects into account have gained increased attention, such as adaptive therapy2 and extinction therapy3.
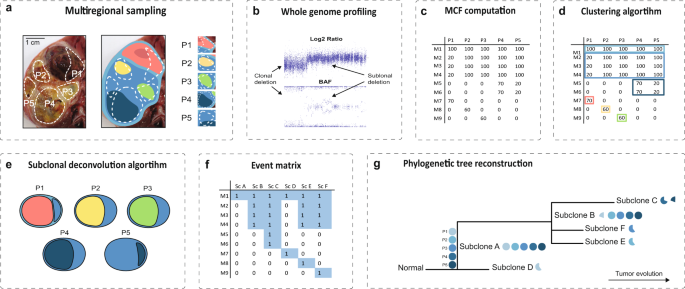
By collecting biopsies from multiple positions in the tumor (multiregional sampling) and at different time points (multitemporal sampling), the evolutionary history of the tumor can be tracked. This analysis is aided by reconstructing so called ancestral (phylogenetic) trees, visualizing the order of genetic alterations as well as which populations of cells are present in the tumor at different positions and time points.
Most current methods to analyze the intratumoral heterogeneity and inferring the genetic history of a tumor are limited to somatic point mutations and does to a large extent disregard copy number variations (an increased or decreased number of copies of a chromosomal segment) or assume that they are clonal (found in all cells). This is a serious shortcoming since most pediatric tumors, as well as high-grade adult tumors show a vast amount of copy number changes, many of which also are subclonal (found in a proportion of cells) in a biopsy. To disregard these chromosomal changes might hamper the inference of the most likely history of the patient’s tumor.
In our newly published article in Communications Biology, we propose DEVOLUTION. It is a tool allowing incorporation of both copy numbers and/or sequencing information for phylogenetic tree inference, provided the proportion of cells that has a certain alteration in a biopsy (the mutated clone fraction, MCF) is known. This allows for phylogenetic trees to be constructed using copy number information alone, sequencing information alone or using both together. DEVOLUTION can also include genetic alterations that are unique for a subset of samples. Many current tools do not allow inclusion of such alterations, which might result in missing information on metastatic-specific changes in the inference process.

Figure 1 An example of a phylogenetic tree produced by DEVOLUTION for a rhabdomyosarcoma (muscle tumor). The filled pies to the left illustrate which biopsies are available from this patient, i.e. 4 biopsies from the primary tumor (B), 3 biopsies from the local relapse (R) and one biopsy from a metastasis (M). The endpoints represent different subclones, whose fraction per sample are visualized by pie charts. The scale bar indicates the distance corresponding to one genetic aberration. Here the relapse and metastasis form their own branch harboring several additional genetic alterations compared with the primary tumor.
In addition, due to intratumoral heterogeneity, each biopsy may contain multiple subclones. This must be taken into account when inferring the possible cell types across the tumor. Not considering this, may result in so called biopsy trees, which are incorrect phylogenies4. DEVOLUTION efficiently resolves subclones and use information from all biopsies during the process to identify which types of cells exist in the tumor and metastases, how they are related, and where they are located. DEVOLUTION could thus be used to investigate how a tumor changes in time during therapy and identify which subgroups of cells constitute relapses and metastases. It could also be used to map the metastatic pathway the cells take to colonize other parts of the body.
In addition, DEVOLUTION allows phylogenies to be produced as part of an objective framework, based on preset rules. This makes it possible to compare trees between patients as well as trees obtained from different studies, without the risk of subjective bias. The program can be accessed through github. A manual for how to use the program along with examples is provided at the github repository.
Github repository: https://github.com/NatalieKAndersson/DEVOLUTION




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in