
Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults and most patients are diagnosed with early stage disease, which is often asymptomatic or weakly symptomatic and doesn’t require treatment at diagnosis. Some patients maintain stable disease which never requires treatment, whilst others develop a progressive disease and develop symptoms that can affect life expectancy. As such, it is clear that not all patients progress at the same rate.
Treatment for CLL has been transformed by the advent of highly effective chemoimmunotherapy regimens and small molecular inhibitors, including those targeted against the B-cell receptor (BCR) which is critical for survival of leukemic lymphocytes. Emerging evidence suggests that early treatment with BCR inhibitors for patients at high-risk of progression could significantly delay the onset of symptomatic leukemia and improve survival. Numerous methods are currently used to determine prognosis, including the Binet disease staging system and the International Prognostic Index, incorporating a combination of clinical, biological and genetic features. For example, unmutated immunoglobulin heavy chain variable region (IGHV) genes or positivity for CD38 surface expression are both associated with progressive disease and poor outcome. Likewise, mutations affecting the TP53, NOTCH1 and SF3B1 genes also drive disease progression and affect patient survival. However, many of these alterations are acquired by the leukemic cells as disease progresses and are relatively uncommon in newly diagnosed CLL. As such, identifying newly-diagnosed patients at high risk of progression and who might benefit from early treatment is very challenging.
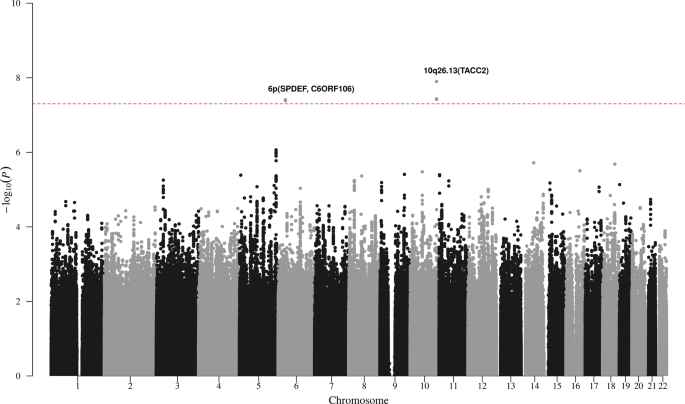
Thanks to a highly collaborative network of CLL researchers and clinicians we established a large multicentre study incorporating 842 CLL patients with long-term follow-up. We looked at over 5 million genetic variants in the human genome and identified two that strongly associated with risk of developing symptomatic CLL requiring treatment. These variants are located on chromosome 6 and chromosome 10, and are carried by 12% and 22% of CLL patients, respectively. Critically, the chromosome 6 and 10 variants are particularly powerful prognostic markers in CLL patients diagnosed with early stage disease and also in patients without high-risk markers typically associated with disease progression. Laboratory tests for genetic markers, such as those identified in our study, are relatively inexpensive and easy to perform, making them ideal for incorporation into routine clinical practise. Therefore, we believe that our findings will allow physicians to more clearly identify CLL patients who might benefit from early treatment before disease progression.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in