
Colorectal cancer (CRC) is the third most common cancer worldwide, with current treatment options involving surgical intervention with chemo- and radiotherapy1. The majority of CRCs display mutations in the tumour suppressor gene adenomatous polyposis coli (APC) - a gene which when suppressed or inactivated leads to constitutively active Wnt/β-catenin signalling, intestinal polyp formation and increased cancer susceptibility2. We therefore wanted to gain a better understanding of the cellular processes that may influence tumour formation following the loss of APC, and importantly identify targets and mechanisms in which tumourigenesis may be suppressed.
Which processes are required for Apc-deficient growth?
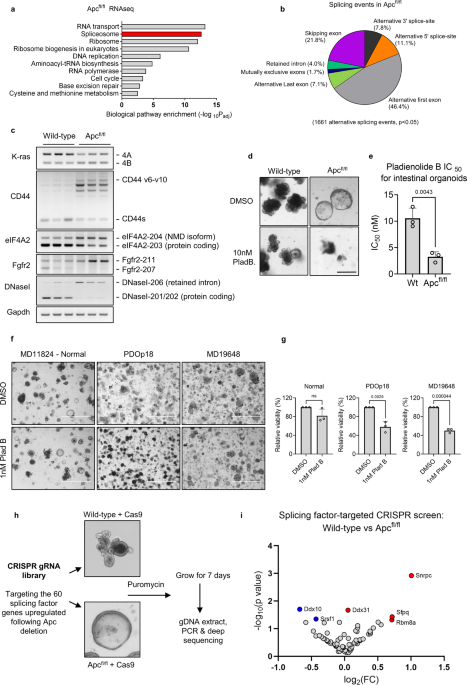
To identify these early cellular processes we began by performing bulk RNA sequencing on intestinal tissue in which Apc had been deleted, and comparing it to normal intestinal tissue. Following pathway analysis, we discovered that the spliceosome, a multi-protein complex that regulates alternative mRNA splicing was significantly enriched. Alternative RNA splicing is a ubiquitous process in which coding and non-coding mRNA sequences are removed and assembled and can form multiple mRNA transcripts from a single pre-mRNA, leading to proteomic diversity3. Given that this process is so well conserved and required for every cell type, we wondered whether the increased activity in Apc-deficient cells could be therapeutically targeted, or whether the inhibition of RNA splicing may cause widespread cellular dysfunction across normal and tumourigenic tissues. Using a previously reported splicing inhibitor, Pladienolide B, (whose derivatives have had some success in haematopoietic cancers driven by splicing factor mutations4) we found that both human and mouse tumour organoids were much more sensitive to splicing inhibition than normal tissue – indicating that splicing may be preferentially targeted in CRC.
To systematically determine if there were genes involved in RNA splicing that may be targeted which were dispensable for normal intestinal homeostasis we developed a synthetic lethal CRISPR/Cas9 screen where a library of guide RNAs targeting the upregulated spliceosome genes following Apc loss were transduced into both wild-type and Apc-deficient organoids. After deep-sequencing we identified a splicing factor, SRSF1, whose guides were significantly depleted in tumour cells compared to normal cells. To validate these observations we performed knock-down experiments in a variety of tumour organoids, with complex oncogenic genetic background and consistently found the loss of Srsf1 significantly inhibited the growth of these tumours, but importantly once again did not affect normal tissue. These findings were mirrored in vivo using a genetic deletion of Srsf1 in combination with Apc in the intestine, demonstrating that not only does Srsf1 regulate proliferation in the intestinal villi, but its loss also resulted in a reduction in stem cell function – indicated through clonogenic assays. Given the importance of functional stem cells (marked by LGR5 expression) in the normal intestinal crypt we also wanted to investigate whether these were effected by Srsf1 loss. Remarkably, using the same clonogenic assays we determined that Srsf1 was dispensable for normal stem cell function.
Can altered splicing affect intestinal cell plasticity?
The next important question was to answer how this was all occurring, and what was the contribution of Srsf1 to these Wnt-driven splicing events. We again performed bulk RNAseq on Apc-deficient tumour tissue, with or without Srsf1 deletion, and performed splicing analysis, and gene-set enrichment analysis. We identified that a significant proportion of splicing changes were dependent on Srsf1, indicating a high requirement for this protein in the dysregulated splicing program following Apc-loss. We also discovered that the loss of Srsf1 promoted an enrichment in genes associated with a more differentiated enterocyte signature, while decreasing stem cell and crypt markers. This was extremely interesting, and led us to ask whether Srsf1 may have a role in stem cell plasticity and in particular de-differentiation. Intestinal cell de-differentiation, is a process in which non-stem cells and differentiated cells can undergo a transition into cancer stem cells. This can be induced through stem cell loss following radiation or chemotherapy, but importantly can be a driver of relapse5,6. To answer whether Srsf1 did in fact regulate intestinal cell plasticity we performed de-differentiation assays in NF-κB and Kras-driven models of CRC with Srsf1 loss. We further validated these findings by performing FACS using a marker of intestinal stem cells (EphB2) where we could separate differentiated cells from stem cells, in a model where we could inducibly deplete both Apc and Srsf1. By performing colonogenic assay we found Srsf1 was indeed a requirement for intestinal cell plasticity.
How does the loss of Srsf1 affect tumour growth and invasion?
Exactly how Srsf1 was regulating this was regulating tumour cell growth was still to be determined. Our results from our RNAseq indicated that the loss of Apc promoted a switch in Kras isoforms to a more pro-tumourigenic version (4B), which was reversed upon Srsf1 loss. We then performed rescue experiments, overexpressing Kras4B in Srsf1 knocked down organoids and found that Kras4B overexpression completely abolished the growth defects found with loss of Srsf1. We further explored this using an invasive, carcinogen-induced model of CRC (p53+AOM), and found Srsf1 led to a significant reduction in invasive tumours, as well as promoting a more differentiated tumour, with lower expression of stem-cell marker PROX1.
Is this clinically relevant?
Using patient-derived organoids, and tissue samples we found that SRSF1 knockdown led to decreased organoid growth across multiple organoid lines, as well as being positively correlated with PROX1 in human TMAs. Importantly the finding that SRSF1 can effect KRAS4B splicing in human tumours encourages the idea of using an available Kras4B inhibitor, deltarasin7, as a therapeutic option in CRC.
What unanswered questions do we still have?
In summary, we have demonstrated that splicing, and in particular splicing factor SRSF1 is absolutely required for the growth of CRC and is an essential regulator of intestinal cell plasticity. Exactly which pathway(s) may be responsible for this, whether KRAS4B is a regulator of plasticity, and exactly what cell population changes occur following Srsf1 loss in the context of de-differentiation are the remaining interesting questions we still have to answer.

References
1. Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71, 209-249, doi:10.3322/caac.21660 (2021).
2. Segditsas, S. & Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 25, 7531-7537, doi:10.1038/sj.onc.1210059 (2006).
3. Wilkinson, M. E., Charenton, C. & Nagai, K. RNA Splicing by the Spliceosome. Annu Rev Biochem 89, 359-388, doi:10.1146/annurev-biochem-091719-064225 (2020).
4. Bonnal, S. C., Lopez-Oreja, I. & Valcarcel, J. Roles and mechanisms of alternative splicing in cancer - implications for care. Nat Rev Clin Oncol 17, 457-474, doi:10.1038/s41571-020-0350-x (2020).
5. de Sousa, E. M. F. & de Sauvage, F. J. Cellular Plasticity in Intestinal Homeostasis and Disease. Cell Stem Cell 24, 54-64, doi:10.1016/j.stem.2018.11.019 (2019).
6. Merlos-Suarez, A. et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 8, 511-524, doi:10.1016/j.stem.2011.02.020 (2011).
7. Zimmermann, G. et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 497, 638-642, doi:10.1038/nature12205 (2013).





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in