STK11 gene (also known as LKB1) codifies for an enzyme called serine/threonine kinase 11. LKB1 is a tumor suppressor helping the cells to not divide so fast and in an uncontrolled way. However, LKB1 is a multitask protein that participates in a number of biological processes relevant in tumor biology, including proliferation, apoptosis, angiogenesis, metabolism and DNA damage (1). Indeed, LKB1 occupies a central position within Kinome dendrogram and it has been described to be a master kinase (2). As it happens with other famous tumor suppressors (TP53, PTEN…etc) most of the knowledge acquired about their role in cancer is based on the loss of function of these proteins, however, somatic missense mutations also occur. STK11 missense mutations represent 30% of all described genetic alterations, and their role in tumor biology is mostly unknown.
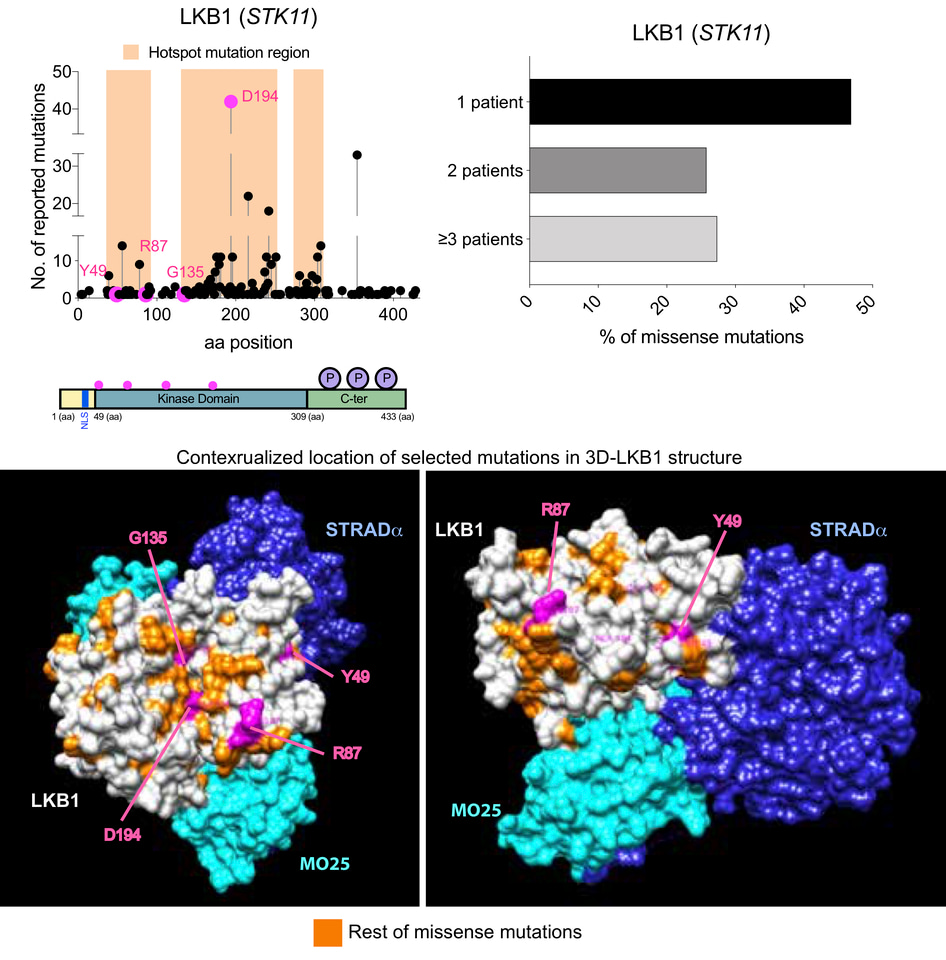
In the late 2000’s we contributed to understand how oncogenes (BRAF) control metabolism of tumor cells through LKB1 (3). Then, in the mid 2010’s we investigated the consequence of LKB1 loss in BRAF mutated lung tumors and discovered a novel role for LKB1 as a UV-induced DNA damage sensor in vivo (4,5), supporting the multitask role of this protein. Now, we have studied the role of four STK11 missense somatic mutations in cancer. Due to the multitask functions of LKB1, these four missense LKB1 somatic mutations were chosen because our interest in studying the different functional parts of the protein contextualized in the human disease. The great majority of missense mutations in STK11, except for D194 residue, do not represent hotspots. D194N mutant was described to be kinase dead. However, the functional consequences in cancer of this mutant were mostly unknown. This residue is located in the ATP binding cleft. G135 residue is also located in the same functional region. Thus, G135R mutant was selected to be compared with D194Y since it was located in a similar 3D functional location (ATP binding cleft). Y49 residue was embedded within a b-sheet in the LKB1 N-lobe close to the 3D interaction region with STRAD. Since LKB1 subcellular localization and kinase activity is STRAD-binding dependent we were interested in studying the possible consequences of this mutation. Finally, R87 is exposed at the surface of the molecule being susceptible to be modified or interact with other molecules. Moreover, the change for the lysine is particularly interesting because t is not very disruptive respect the charge, however Lys and Arg have different H-bonding capabilities and hydration free energies, and they can be post-translationally modified differently. Importantly, all selected mutations were initially detected in tumor samples and were predicted to be oncogenic or likely oncogenic (polyphen or OncoKB team TCGA).

Figure 1. Graph showing the number of STK11 reported mutations in lung cancer, melanoma and cervix (COSMIC and TCGA) . Below, LKB1 primary structure showing in pink the location of the mutations studied in the investigation. On the right, graph shows the percentage of mutations identified in one, two or in three or more patients. Bottom, 3D-LKB1-MO25-STRADa structure (2WTK.pdb) showing the relative position of the mutants in the 3D structure.
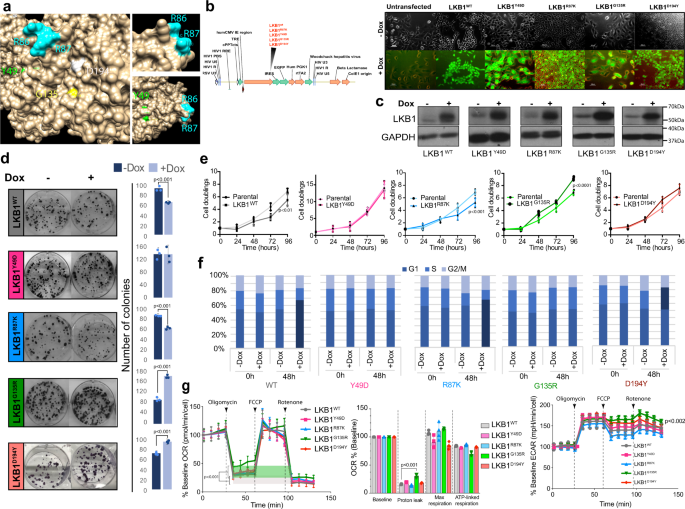
In cancer, contributing mutations are selected according to the particular mutational context. Thus, the election of an appropriate system to study the functional contributions of a mutated protein compared to its wild type counterpart, is always challenging. To establish different phenotypic tumor suppressor capabilities of the mutant isoforms, we used three different LKB1-null cell lines (lung cancer, melanoma and cervix). To focus on the molecular changes clinically relevant, we filtered the expression profiles promoted by the wild type isoform with publicly available datasets and robust gene expression signatures for LKB1-deficient tumors. The resultant signature was investigated in every mutant at the expression level and at the protein level (Secretome). Finally, the functional and phenotypical consequences promoted by the mutant isoforms were investigated in vitro and in vivo. One interesting result is that LKB1 missense somatic mutations might contribute to tumor development and/or progression without affecting the tumor suppressor activity (i.e. promoting motility). Other mutants not only loss they tumor suppressor capability but promoted tumor growth, and in another case, the isoform dysregulated the cytokine expression profile further than the loss of LKB1, promoting inflammation in vivo, which might be important for immunotherapy issues. Interestingly, some of these features appear to be independent of the kinase activity of LKB1.
Altogether, our results highlight the relevance of study the missense mutations of tumor suppressor proteins (TP53, STK11, PTEN, RB…etc) further than the loss of the tumor suppressor function, particularly in proteins with a described multitask capabilities. The accumulation if this type of data will increase our knowledge of this disease and will help to manage patients.
Refences
- Gan, R. Y., and Li, H. B. (2014) Recent progress on liver kinase B1 (LKB1): expression, regulation, downstream signaling and cancer suppressive function. Int J Mol Sci 15, 16698-16718
- Lizcano, J. M., Goransson, O., Toth, R., Deak, M., Morrice, N. A., Boudeau, J., Hawley, S. A., Udd, L., Makela, T. P., Hardie, D. G., and Alessi, D. R. (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 23, 833-843
- Esteve-Puig, R., Canals, F., Colome, N., Merlino, G., and Recio, J. A. (2009) Uncoupling of the LKB1-AMPKalpha energy sensor pathway by growth factors and oncogenic BRAF. PLoS One 4, e4771
- Esteve-Puig, R., Gil, R., Gonzalez-Sanchez, E., Bech-Serra, J. J., Grueso, J., Hernandez-Losa, J., Moline, T., Canals, F., Ferrer, B., Cortes, J., Bastian, B., Ramon, Y. C. S., Martin-Caballero, J., Flores, J. M., Vivancos, A., Garcia-Patos, V., and Recio, J. A. (2014) A mouse model uncovers LKB1 as an UVB-induced DNA damage sensor mediating CDKN1A (p21WAF1/CIP1) degradation. PLoS Genet 10, e1004721
- Gonzalez-Sanchez, E., Martin-Caballero, J., Flores, J. M., Hernandez-Losa, J., Ma Angeles, M., Cortes, J., Mares, R., Barbacid, M., and Recio, J. A. (2013) Lkb1 loss promotes tumor progression of BRAF(V600E)-induced lung adenomas. PLoS One 8, e66933




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in