
Estruch M, Reckzeh K et al. Leukemia (2020)
Mutational activation of PI3K/AKT downstream signaling is a common theme in cancer, which promotes survival, DNA-repair, and ultimately tolerance to DNA damaging radiation- and chemotherapy (1). AKT downstream signaling can also be activated by the DNA-dependent protein kinase (DNA-PK), which regulates repair of therapy-induced DNA double-strand breaks through non-homologous end-joining (2). Consistently, therapeutic inhibitors of either PI3K/AKT or DNA-PK sensitize cancer to DNA damaging therapy, highlighting that DNA repair mediated by mutational and/or therapy-induced activation of AKT downstream signaling stands out as a key mechanism of resistance toward DNA damaging therapy.
More than 60% of patients with acute myeloid leukemia (AML) harbor mutations in receptor tyrosine kinases (KIT, FLT3), intracellular kinases (JAK2), or alternative oncogenic driver aberrations such as RAS, which partially confer down-stream activation of the PI3K/AKT signaling pathway (3-7).
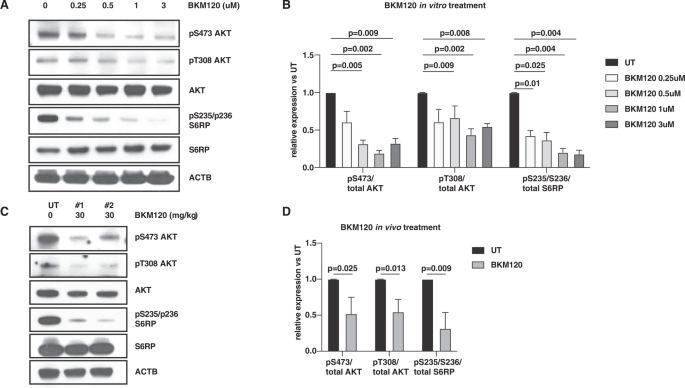
Herein, we demonstrated that mutational PI3K/AKT signaling in a inv(16)/KITD816Y AML mouse model is markedly enhanced by chemotherapy-induced DNA damage through complementary DNA-PK-dependent activation of AKT downstream signaling. Therapeutic inhibition of either PI3K or DNA-PK markedly reduced chemotherapy-induced AKT downstream signaling leading to increased DNA-damage and apoptosis of inv(16)/KITD816Y AML cells treated with conventional AML chemotherapy. Consistently, combinatorial treatment with chemotherapy and a PI3K inhibitor or a DNA-PK inhibitor synergistically inhibited in vitro growth and survival of AML cells, and significantly prolonged survival of inv(16)/KITD816Y AML mice compared to untreated/single treated mice.
Conceptually, our study implicates that constitutive mutational PI3K/AKT downstream signaling cooperates with chemotherapy-induced DNA-PK-dependent enhancement of AKT downstream signaling to promote survival, DNA repair, and chemotherapy resistance. Thus, our study provides experimental evidence for combining chemotherapy with inhibitors of DNA-PK, PI3K, or alternatively relevant inhibitors of upstream PI3K/AKT activators such as mutant KIT and mutant FLT3, in order to enhance chemotherapy response and overcome resistance. Our study also emphasizes that such inhibitors of AKT downstream signaling (i.e. DNA-PKi, PI3Ki, or KITi and FLT3i) should be administered concurrently with chemotherapy rather than sequentially to potentiate rather than complement chemotherapy response as is the case for current therapeutic regimens combining AML chemotherapy with the FLT3 inhibitors midostaurin or quizartinib (8, 9). Indeed, this is supported by clinical data demonstrating that concomitant treatment of patients with relapsed or refractory chronic lymphatic leukemia with a PI3K inhibitor (idelalisib) and antibody/chemotherapy almost doubled median progression-free survival at expense of somewhat augmented toxicity compared to treatment with antibody/chemotherapy alone (20.8 vs 11.1 months, hazard ratio 0.33, 95% CI 0.25–0.44; p < 0.0001)(10).
Hence, our study provides a strong rationale for future clinical studies selecting AML patients exhibiting constitutive mutational PI3K/AKT activation for simultaneous treatment with chemotherapy and inhibitors of DNA-PK, PI3K, or relevant upstream activators such as mutant KIT, FLT3, JAK2, and RAS to enhance chemotherapy response. In a broader perspective, our study highlights the potential of novel AML treatment modalities combining standard chemotherapy with simultaneous targeted inhibition of key signaling nodes at the interface of oncogenic signaling and DNA damage response pathways to overcome chemotherapy resistance and ultimately improve clinical outcome of AML patients.
References
- Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin Cancer Biol. 2019;59:125-32.
- Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Molecular cell. 2008;30(2):203-13.
- Rapin N, Bagger FO, Jendholm J, Mora-Jensen H, Krogh A, Kohlmann A, et al. Comparing cancer vs normal gene expression profiles identifies new disease entities and common transcriptional programs in AML patients. Blood. 2014;123(6):894-904.
- Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102(3):972-80.
- Pardee TS, Zuber J, Lowe SW. Flt3-ITD alters chemotherapy response in vitro and in vivo in a p53-dependent manner. Exp Hematol. 2011;39(4):473-85 e4.
- Dos Santos C, McDonald T, Ho YW, Liu H, Lin A, Forman SJ, et al. The Src and c-Kit kinase inhibitor dasatinib enhances p53-mediated targeting of human acute myeloid leukemia stem cells by chemotherapeutic agents. Blood. 2013;122(11):1900-13.
- Xiao Y, Deng T, Su C, Shang Z. MicroRNA 217 inhibits cell proliferation and enhances chemosensitivity to doxorubicin in acute myeloid leukemia by targeting KRAS. Oncol Lett. 2017;13(6):4986-94.
- Stone RM, Larson RA, Dohner H. Midostaurin in FLT3-Mutated Acute Myeloid Leukemia. The New England journal of medicine. 2017;377(19):1903.
- Altman JK, Foran JM, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213-21.
- Zelenetz AD, Barrientos JC, Brown JR, Coiffier B, Delgado J, Egyed M, et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017;18(3):297-311.
Figure Caption: Rationale for concomitant treatment of oncogene-mutated AML patients with chemotherapy and either PI3K or DNA-PK inhibitors. A) Inv(16)/KITmut AML exhibits constitutive activation of the PI3K/AKT signaling pathway, promoting survival and DNA repair of AML cells. B) The chemotherapeutic Doxorubicin (DOX) induces double strand brakes (DSBs) and activation of a DNA damage response (DDR) partially through DNA-PK-dependent complementary AKT activation of inv(16)/KITmut AML, which enhances DNA repair, survival and therapy resistance of AML cells. C) and D) Concomitant treatment with chemotherapy and an inhibitor of PI3K (C) or an inhibitor of DNA-PK (D), impairs AKT downstream signaling, resulting in enhancement apoptosis and improved survival of inv(16)/KITmut AML.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in