
For patients with early stage breast cancer, the treatment outcome could be quite different. Some patients are essentially cured after the surgery to remove the primary tumor and chemotherapy to prevent relapse. On the other hand, some other patients develop recurrent diseases, often as metastatic lesions in distant vital organs. These metastatic cancers usually respond poorly to standard treatments, eventually leading to the death of the patients. With the development of gene expression profiling technology, gene signatures have been defined to identify patients with poor prognosis breast cancer, and such signatures have been applied in clinical practice to help inform treatment decisions. However, the driver genes behind such aggressive cancers remained largely unknown. Without such knowledge, options to prevent or effectively control metastatic breast cancer are limited.
A common theme in the identification of genetic drivers of cancer is that such important genes are often affected by recurrent genomic alterations, such as BCR-ABL fusion oncogene produced by genomic translocation in CML and Her2 amplification in Her2-positive breast cancer. We developed a bioinformatic method to mathematically convert publicly available mRNA gene expression data of clinical breast cancer data sets into predicted recurrent genomic amplifications or deletions associated with poor prognosis breast cancer1. Using this approach, we identified and validated (using FISH) a chromosome location (8q22) that is frequently amplified in poor prognosis tumors. In this locus, we further identified MTDH as a possible driver gene for aggressive breast cancer. Immunohistochemical analyses showed that MTDH is overexpressed in 30-40% of breast cancer patients, and its high expression level represents substantial higher risk (hazard ratio > 3) for patients to develop metastatic recurrence that leads to shorter survival1. Initial testing using xenograft models showed a role of MTDH in promoting lung metastasis and broad-spectrum chemoresistance of breast cancer.
Prior to this study, there was little knowledge about the functional importance of MTDH in normal physiology or cancer. One previous report suggested that a peptide domain of MTDH is displayed on cell surface and promotes tumor cell adhesion to blood vessels. However, in most normal or cancer cells, MTDH is mostly localized in the cytoplasm, not on the cytoplasmic membrane to mediate such cell-cell adhesion. To investigate the role of MTDH in normal development and validate the functional importance of MTDH in breast cancer progression and metastasis, we generated MTDH whole-body knockout mice. Interestingly, although MTDH is universally expressed in many tissues, Mtdh knockout did not impair mouse development and normal homeostasis. In contrast, Mtdh knockout significantly delayed the breast cancer initiation, progression and metastasis in MMTV-PyMT, MMTV-Neu, MMTV-Wnt and carcinogen-induced breast cancer models2. Similar observation was made when we crossed Mtdh knockout with genetically modified mouse models of prostate, lung, and intestinal cancers3,4. Notably, MMTV-MTDH transgenic mice did not developed spontaneous mammary gland tumors, although transgenic expression of MTDH in mouse mammary epithelial cells at least partially rescued the tumor development defects of Mtdh knockout2. Detailed analyses further revealed that MTDH loss does not affect the activity of normal mammary gland stem cells, but renders mammary tumor initiating cells susceptible to oncogenic stress and chemotherapies, thereby limiting the initiation and expansion of mammary tumors. These findings established MTDH as a classical example of “cancer fitness genes”5,6. Such genes are not crucial for normal development of homeostasis in normal conditions, cannot initiate tumors on its on as an classical oncogene often does in transgenic mouse models, but is indispensable for the initiation, expansion and progression tumors in the face of constant high level of stresses. Such cancer fitness genes represent ideal therapeutic targets, as blocking such genes is likely to cause tumors to collapse under stress, but will spare normal cells under physiological conditions. Indeed, in our recently published studies in Nature Cancer, conditional knockout of MTDH in well-developed mammary gland tumors inhibited tumor progression and lung metastasis, validating MTDH as a suitable therapeutic target even in late stage cancers.
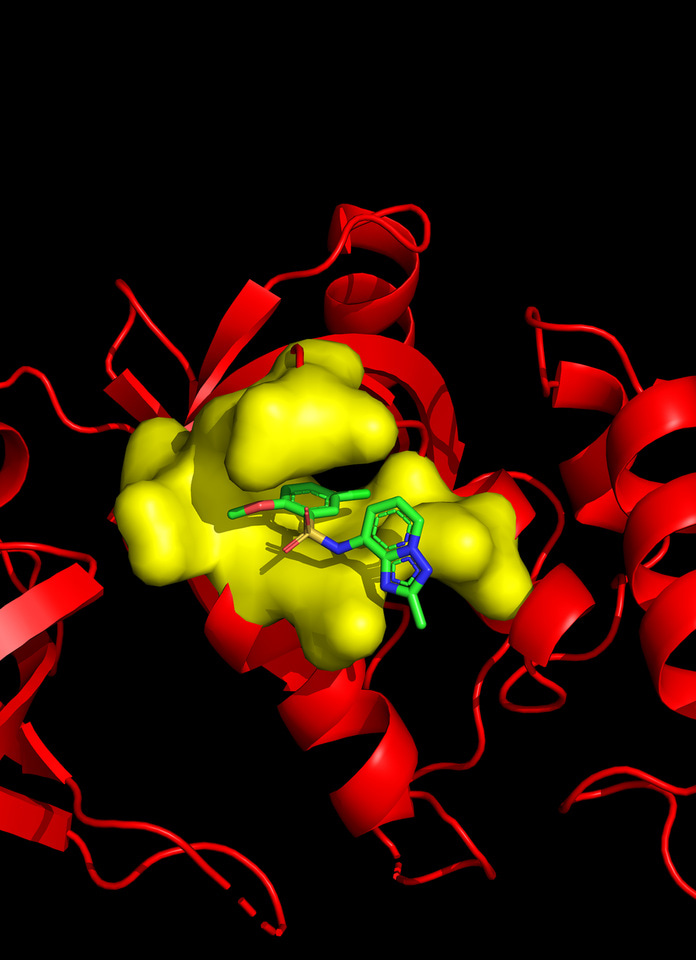
As an important step toward understanding the molecular mechanism of MTDH function, we identified Staphylococcal nuclease domain-containing 1 (SND1) as a key interacting functional partner of MTDH by using co-immunoprecipitation followed by mass spectrometry in 20117. We further resolved the crystal structure of MTDH-SND1 complex through a collaboration with structural biologist Dr. Yongna Xing at the University of Wisconsin–Madison8. The surface contour of SND1 revealed two deep pockets that specifically interact with the MTDH residues. In particular, the hydrophobic side chains of MTDH W394 and W401 were found to be buried deep into the two binding pockets on the surface of SND1. Point mutations of these two Tryptophan residues in MTDH eliminated its interaction with SND1 as well as its tumor promoting functions. Notably, the two binding pockets possess ideal structural, geometrical and biochemical properties suitable for the development of small molecule inhibitors.

With the support from Dr. Hahn Kim at the Small Molecule Screening Center of Princeton, we established a split-luciferase high-throughput screening platform and used it to screen a 50K high diversity singleton chemical library and identified the C26 series compounds that specifically block MTDH-SND1 interaction. The co-crystallization analysis confirmed that C26s indeed compete with MTDH to occupy the W401 pocket on SND1 (Figure 1). Treatment of mice using the C26-A6 compound significantly suppressed breast cancer progression and metastasis, and prolonged the survival of animals.
The development of conditional knockout of MTDH and pharmacological compounds that block MTDH-SND1 function gave us long-awaited tools to evaluate the molecular pathways affected by acute disruption of MTDH in tumors. Consistent with our previous studies indicating that MTDH-SND1 enhances the survival of tumor cells under chemo-induced stresses, we observe negative enrichment of cell growth and survival genes and pathways after acute MTDH disruption. Some of the key gene changes include up-regulation of Cdc20, Plk1 and Myc. Another prominent change is the activation of Interferon pathways upon MTDH inhibition. This is consistent with our earlier observation that the deficiency of Mtdh KO tumors in developing lung metastasis as compared to MTDH wild-type cells is much more prominent in immunocompetent mice than in immunodeficient hosts. We therefore speculated that in addition to enhancing tumor intrinsic survival, MTDH enable evasion of tumor cells from immunosurveillance. Detailed mechanistic studies revealed that MTDH-SND1 complex reduces tumor antigen presentation by promoting RNA degradation of the key antigen processing and presentation machinery proteins Tap1 and Tap2. C26-A6 treatment restores anti-tumor immunosurveillance to inhibit breast cancer progression and metastasis, and synergizes with immune checkpoint blockade therapy to induce regression or stabilization of late stage metastatic breast cancer in mouse models.

Figure 2. Milestones in identifying MTDH as a key cancer fitness gene and developing MTDH-targeting therapy.
The discovery, molecular characterization, and therapeutic targeting of MTDH is a long journey that took more than 15 years to date (Figure 2) and represents a multidisciplinary effort combing expertise in diverse fields of bioinformatics, cancer biology, genetics, biochemistry, structural biology, medicinal chemistry, pathology, and stem cell biology. Our studies pave the way toward targeting MTDH in a wide spectrum of major human malignancies. The genetic and pharmacological tools we established in these studies will enable us to tackle the next set of questions regarding the biochemical mechanism of the MTDH-SND1 complex, its normal physiological functions, and its multifaceted roles in cancer progression (Figure 3).

References:
1 Hu, G. et al. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer cell 15, 9-20 (2009).
2 Wan, L. et al. MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer cell 26, 92-105 (2014).
3 Wan, L. et al. Genetic ablation of metadherin inhibits autochthonous prostate cancer progression and metastasis. Cancer research 74, 5336-5347, doi:10.1158/0008-5472.CAN-14-1349 (2014).
4 Shen, M. et al. Therapeutic Targeting of Metadherin Suppresses Colorectal and Lung Cancer Progression and Metastasis. Cancer research 81, 1014-1025, doi:10.1158/0008-5472.CAN-20-1876 (2021).
5 Shen, M. & Kang, Y. Stresses in the metastatic cascade: molecular mechanisms and therapeutic opportunities. Genes Dev 34, 1577-1598, doi:10.1101/gad.343251.120 (2020).
6 Esposito, M., Ganesan, S. & Kang, Y. Emerging strategies for treating metastasis. Nat Cancer 2, 258-270, doi:10.1038/s43018-021-00181-0 (2021).
7 Blanco, M. A. et al. Identification of staphylococcal nuclease domain-containing 1 (SND1) as a Metadherin-interacting protein with metastasis-promoting functions. The Journal of biological chemistry 286, 19982-19992 (2011).
8 Guo, F. et al. Structural insights into the tumor-promoting function of the MTDH-SND1 complex. Cell reports 8, 1704-1713 (2014).



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in