Shiva Bamezai, Christian Buske and Vijay P. S. Rawat
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive leukemia predominately found in children, is associated with inferior survival outcomes in adults and shows an average survival of only 35%. T-ALL patients are treated with high-dose multi-agent chemotherapy that is often associated with long-term side effects. High-risk T-ALL patients are generally prioritized for hematopoietic stem cell (HSC) transplantation, however, the clinical outcome of primary resistant leukemias remains extremely poor and relapse of the disease is the most common cause of treatment failure. Therefore, we require a better understanding of the biology of this disease to identify novel drug targets to improve treatment outcomes. We at the Institute for Experimental Cancer Research at the University of Ulm, Germany, focus on characterizing the role of genetic alterations and aberrant expression of embryonic factors in the pathogenesis of acute leukemia1-3. In this study, we report that the embryonic, dioxygenase enzyme Ten eleven translocation1 (TET1) is aberrantly overexpressed in T-ALL patients, plays an essential pro-leukemic role in T-ALL and is a potential novel drug target in this disease.
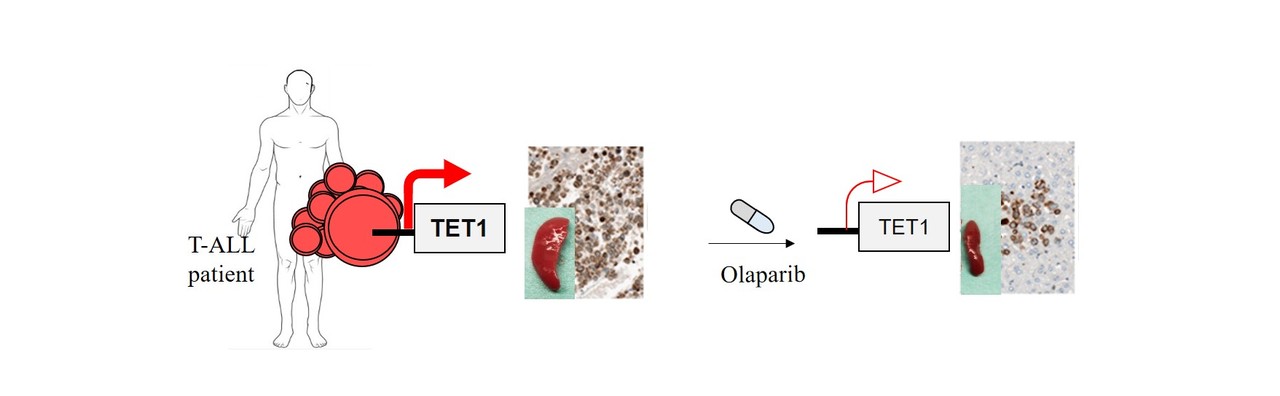
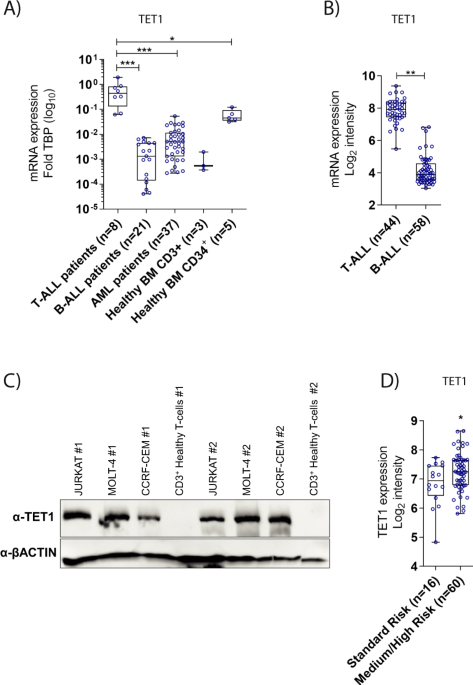
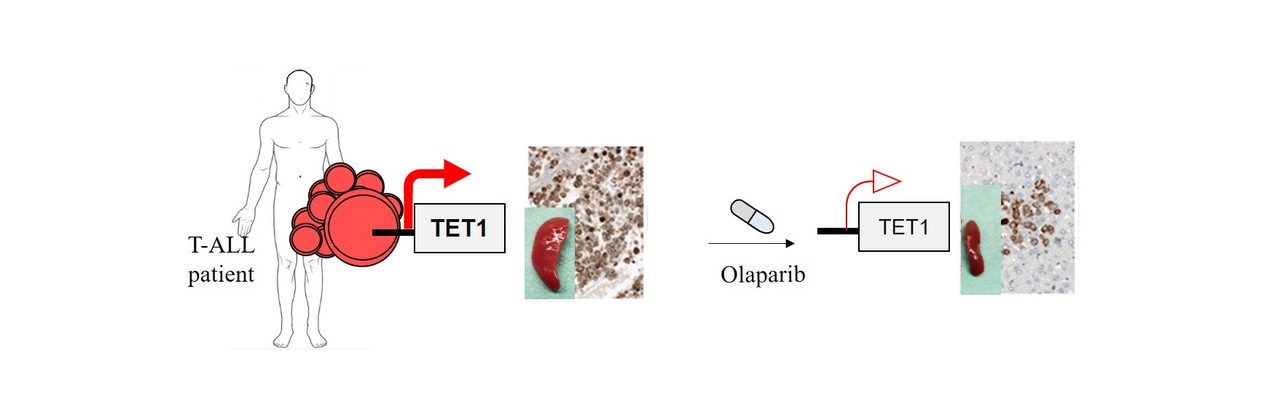
TET1 is a founding member of the TET family of epigenetic enyzmes that establish 5-hydroxymethylcytosine (5hmC) marks on DNA via the oxidation of 5-methylcytosine4, 5. TET and 5hmC play a vital role in active gene regulation, maintenance of genome integrity, normal stem self-renewal, differentiation and their dysfunction has been associated with cancer 6-8. Although TET genes are described as tumor suppressors and TET1 is classified as a tumour suppressor of haematological neoplasms, especially in B-cell malignancies 6, to our surprise our team observed based on data of three independent study groups that TET1 is highest expressed in T-ALL patients compared to other leukemia types and aberrantly overexpressed in the large majority of patients compared to their healthy counterparts. Stable knockdown, CRISPR-Cas9 mediated knockout and overexpression strategies revealed that T-ALL cells require high TET1 expression for their leukemogenic growth. Overexpression of the TET1 catalytic domain was sufficient to augment leukemic growth of T-ALL cells in xenografts. In contrast to its growth promoting effect in T-ALL, TET1 was dispensable for the development of healthy thymic progenitors and functionally mature T cells. Next generation sequencing of RNA and global 5hmC marks, accompanied by gene expression analysis of T-ALL patients indicated that TET1 promotes leukemic growth by maintaining the expression of T-ALL specific oncogenes, cell cycle and DNA repair pathway genes via establishment of 5hmC marks at their promoters and gene bodies. The relevance of high TET1 expression for the maintenance of genome integrity of T-ALL cells was further evidenced by gross DNA damage in primary leukemia cells and human cell lines deprived of TET1 expression, which rendered the leukemic cells vulnerable to DNA damaging radiation. Taken together, these data point to dichotomous functions of TET1 in acute lymphoid leukemia, uncover an unexpected pro-leukemic role and suggest that T-ALL cells hijack the DNA repair function of TET1.
In addition, we identified single stranded DNA repair associated enzymes of the PARP family as being highly expressed in T-ALL, acting as upstream regulators of TET1. This allowed us to use the FDA approved PARP inhibitor, Olaparib to abrogate TET1 expression and by this to induce global loss of 5hmC marks and to antagonize leukemic growth of T-ALL cells in vivo. These data imply that PARP inhibitors can be a potentially additional therapeutic tool beside conventional chemotherapy in the treatment of T-ALL patients. In the future it would be interesting to check whether TET1 expression level can be an important determinant of Olaparib sensitivity in cancer therapy.

1. Rawat VP, Cusan M, Deshpande A, Hiddemann W, Quintanilla-Martinez L, Humphries RK, et al. Ectopic expression of the homeobox gene Cdx2 is the transforming event in a mouse model of t(12;13)(p13;q12) acute myeloid leukemia. Proc Natl Acad Sci U S A 2004 Jan 20; 101(3): 817-822.
2. Rawat VP, Arseni N, Ahmed F, Mulaw MA, Thoene S, Heilmeier B, et al. The vent-like homeobox gene VENTX promotes human myeloid differentiation and is highly expressed in acute myeloid leukemia. Proc Natl Acad Sci U S A 2010 Sep 28; 107(39): 16946-16951.
3. Rawat VP, Thoene S, Naidu VM, Arseni N, Heilmeier B, Metzeler K, et al. Overexpression of CDX2 perturbs HOX gene expression in murine progenitors depending on its N-terminal domain and is closely correlated with deregulated HOX gene expression in human acute myeloid leukemia. Blood 2008 Jan 1; 111(1): 309-319.
4. Khoueiry R, Sohni A, Thienpont B, Luo X, Velde JV, Bartoccetti M, et al. Lineage-specific functions of TET1 in the postimplantation mouse embryo. Nat Genet 2017 Jul; 49(7): 1061-1072.
5. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009 May 15; 324(5929): 930-935.
6. Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol 2015 Jun; 16(6): 653-662.
7. Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I, et al. 5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability. Cell Rep 2016 Feb 16; 14(6): 1283-1292.
8. Zhong J, Li X, Cai W, Wang Y, Dong S, Yang J, et al. TET1 modulates H4K16 acetylation by controlling auto-acetylation of hMOF to affect gene regulation and DNA repair function. Nucleic Acids Res 2017 Jan 25; 45(2): 672-684.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in