

Subsets of T-cell acute lymphoblastic leukemias are characterized by their aberrant expression of transcription factors such as TAL1, TLX1, and TLX3. This often results from chromosomal translocations that place gene regulatory elements upstream of the transcription factor to drive uncontrolled expression. However, high transcription factor levels alone are insufficient to transform normal blood cells, rather additional mutations are required to promote leukemia.
Our long-term interest involves understanding how co-occurring mutations might cooperate to initiate and drive T-cell acute lymphoblastic leukemia. In earlier work we showed that JAK3 mutations are more likely to be present in T-ALL patients that overexpress the transcription factor HOXA91. Now as next-generation sequencing data becomes available for large patient cohorts, new associations between different mutations are being unveiled.
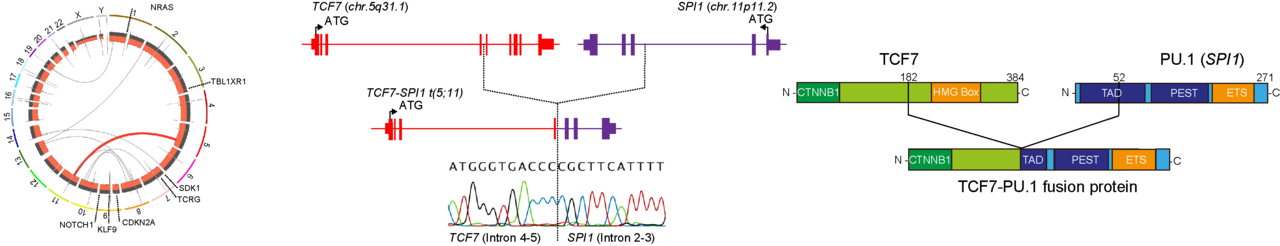
Using both whole-genome and RNA-sequencing we originally identified a single patient with a novel TCF7-SPI1 fusion2. The patient’s T-ALL cells also showed additional mutations in NRAS, NOTCH1, KLF9, CDKN2A as well as other genes. Afterwards a publication appeared from the laboratory of Junko Takita showing SPI1 gene fusions in 4% of T-ALL patients3. Closer analysis of the Japanese cohort indicated the fusion-positive cases often had co-occurring mutations in NRAS similar to our case study. Following up on this important clue, we focused on determining if the expression of both NRAS(G12D) and TCF7-SPI1impacted disease development using mouse models.
One of the challenges in studying oncogenic cooperation in vivo is to maintain the correct cellular context. This means that oncogenes should remain restricted to developing T-cells. This was particularly important here given the strong oncogenic potential of activating NRAS mutations. Rather than following the conditional transgenic mouse model route to achieve restricted expression, we used our newly developed retroviral vector system that has asymmetric lox66/71 sites to irreversibly ‘flip’ our genes of interest into the correct orientation in the presence of Cre-recombinase (Figure 1A). Using a bone marrow transplant model with hematopoietic progenitor cells isolated from CD2-Cre transgenic donor mice, we were then able to drive expression of NRAS(G12D) and TCF7-SPI1 alone or together within developing T cells (Figure 1B). Importantly this showed that only the combination resulted in the development of an immature T-ALL.

We complemented these findings with single-cell analysis of TCF7-SPI1 fusion-positive and negative patients. The fusion-positive cases showed enrichment of Wnt/β-catenin signaling which highlighted the potential importance of the β-catenin binding domain at the N terminus of the fusion. Indeed, deletion of this domain altered the phenotype of the disease to that caused by expression of NRAS(G12D) alone. Likewise, pharmacological antagonism of the TCF/β-catenin interaction using PKF 118-310 was able to inhibit the transcriptional activity of the fusion. Notably treatment PKF 118-310 of PDX cells and in vivo genetic knockdown studies showed that loss of the fusion resulted in leukemia cell differentiation. Moreover, using an in vivo limiting dilution assay we demonstrated that the presence of the fusion led to a higher frequency of leukemic stem cells. Taken together, the TCF7-SPI1 fusion leads to a more immature disease with higher leukemic stem cell frequency. From the translational perspective, targeting the fusion with β-catenin inhibitors can reverse their dedifferentiated state. However, while our data suggests that such therapy against TCF7-SPI1 positive cases would be highly effective in clearing leukemic stem cells via cellular differentiation, this application awaits the arrival of a more clinically appliable antagonists.
Notwithstanding this limitation, some key questions remain for patients that harbor SPI1 translocations. Is a mutant kinase always needed even in those fusion-positive cases that do not have an activating RASmutation? Does the Wnt/β-catenin pathway play a role for alternate N-terminal fusion partners such as STMN1-SPI1? The answer to the first question has now been provided from paper published during the revision period of our manuscript4. Here, they demonstrated that TCF7-SPI1 fusion positive cases are exquisitely sensitive to dasatinib, thus providing some evidence that activated kinase signaling is absolutely required in these leukemias

Right hand side: Lab of Dr Charles de Bock (Top right) alongside Sofia Omari (Research Officer, top left), Jackie Huang (Post-doctoral Scientist, Bottom left) and Tommy Seo (Honours Student, Bottom Right) having a lab meeting online due to COVID driven lockdown.
This work was carried out by an international collaboration, including the laboratories led by Professor Jan Cools at KU Leuven, Belgium and Dr. Charles de Bock at the Children’s Cancer Institute, UNSW, Sydney, Australia. However, like many laboratories around the world, our ability to undertake important experimental revisions was also challenged by the ongoing COVID-19 situation. We therefore thank the many co-authors who worked tirelessly during this period in helping us strengthen this paper and also the editorial team of Nature Communications who were very supportive throughout the process.
References
- de Bock, C. E. et al. HOXA9 cooperates with activated JAK/STAT signaling to drive leukemia development. Cancer Discov. 8, 616–631 (2018).
- De Bie, J. et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. 1358–1369 (2018) doi:10.1038/s41375-018-0127-8.
- Seki, M. et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. (2017) doi:10.1038/ng.3900.
- Gocho, Y. et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat. Cancer (2021) doi:10.1038/s43018-020-00167-4.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in