
Elimination of cancer cells by effector immune cells represents the culmination of a complex cascade of events, and disruption of any of those events may result in resistance. T cell-engaging therapies, such as T cell bispecific antibodies (TCBs) or chimeric antigen receptors (CARs), are raising extraordinary expectations and have been recently approved to treat some hematologic malignancies. In contrast, TCBs and CARs against solid tumors tested to date, have failed to show clinical efficacy. This failure prompted intense research and different strategies are being implemented to overcome primary or acquired resistance mechanisms. All these mechanisms impinge on the ability of T cells to reach cancer cells and/or on the inhibition of T cells. However, little is known about putative intrinsic mechanisms of resistance of cancer cells. That is, mechanisms deployed by tumor cells to resist killing by active and correctly engaged T cells. One of the main interests of our laboratory is to understand the complex biology of tumor cells and immunotherapy, to design novel therapies and to anticipate mechanisms of resistance with the ultimate aim to improve current treatments.

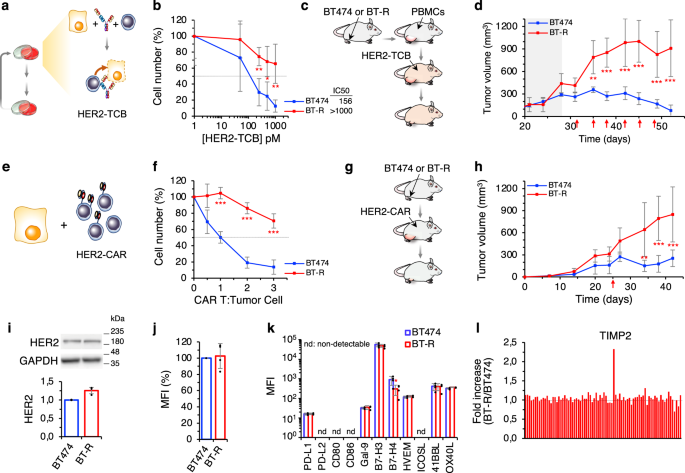
In order to identify unknown mechanisms of resistance to T cell redirection therapies, we started this project 3 years ago. To do so, the first step was to generate a proper model of resistance to T cell based therapies. We chose as a model a HER2+ breast cancer cell line, the BT474, and a TCB targeting HER2. HER2 is a driver for breast and gastric cancers, among other cancers. Its downregulation may lead to tumor regression; thus, HER2 driven cancer cells are not likely to downmodulate its expression. This made them a useful tool to unveil novel mechanisms of resistance. Therefore, we started generating the model by treating repeatedly our cancer cells in vitro with peripheral blood mononuclear cells (PBMCs) from healthy donors and the TCB. Unfortunately, we found that after some rounds of treatment, epithelial cells were lost due to colonization of the plate by remaining PBMCs. To solve this issue, we infected our cells with a virus carrying a resistance gene to an antibiotic, and after each PBMCs and TCB round of treatment, we treated our cells with the antibiotic to kill the remaining PBMCs.
Successfully, after months of treatments, we obtained our resistant model. Then, the most challenging part showed up, to elucidate whether the resistance phenotype was due to an extrinsic mechanism displayed by the T cells, or, by contrast, it was due to an intrinsic mechanism of cancer cells. As stated previously, HER2 was not likely to be downmodulated, and resistant cells maintain the levels of antigen expression. Then, we assessed several markers of activation, proliferation, or secreted cytokines by the lymphocytes, but no changes were observed. In addition, resistant cells did not upregulate any known immune check point inhibitor.
Therefore, we focused on possible mechanisms of intrinsic resistance. We performed a RNA-seq analysis, which evidenced the huge transcriptomic change that the chronic treatment caused in resistant cells. We spent several months trying, and failed to functionally validate different top candidates by loss of function approaches in order to resensitize resistant cells. However, recent works published in high impact journals persuaded us to focus on the interferon-gamma (IFN-gamma) gene set enrichment signature (GSEA). Indeed, we verified that our resistant cells exhibit a striking deficient response to IFN-gamma, even though the gene signature was not among the TOP 10 differentially expressed between parental and resistant cells. The project’s turning point appeared when we treated our cocultures with an IFN-gamma blocking antibody. For the first time we achieved to prevent lymphocyte killing in sensitive cells. This first evidence was corroborated across different breast cancer HER2+ models. To confirm if this phenotype was due to an intrinsic mechanism deployed by tumor cells, as a proof of concept we knocked-out the interferon gamma receptor 1 (IFNGR1) in parental BT474 cells. Confirming our hypothesis, cells without this gene, and consequently deficient in the IFN-gamma signaling pathway, were resistant to TCBs and CARs. This hypothesis was further confirmed in HER2+ lung and ovarian cancer models.
That promising result motivated us to further investigate, and to take a deeper look, at the IFN-gamma signature in the RNA-seq. That’s how we identified JAK2 downmodulation, and its functional validation evidenced its role in resistance to T cell redirection therapies by disrupting the IFN-gamma pathway. We further validated this key finding in two different acquired resistant models generated in vivo. Next, we generated resistant models to the IFN-gamma cytokine and, fully supporting our findings on the critical role of IFN-gamma, resistant cells to IFN-gamma were also resistant to TCBs and CARs. Furthermore, JAK2 loss was confirmed as the mechanism of resistance to IFN-gamma.
Additionally, our research center, the Vall d’ Hebron Institute of Oncology (VHIO), gives us the chance to generate patient’s derived xenografts (PDXs)—which are excellent models that recapitulate the heterogeneity of the original tumor—due to the fluent communication with the Vall d’Hebron Hospital. We used three PDXs in this project to further validate our findings: the IFN-gamma deficient response through JAK2 downmodulation as the mechanism of acquired resistance.
Our results unveil a novel mechanism of resistance to T-cell based therapies, and imply the potential use of JAK2 and IFN-gamma response as a surrogate biomarker of response to immunotherapies. In addition, they open the avenue for the screening for therapies that can overcome deficient IFN-gamma response or restore JAK2 levels. These are the next projects we are starting in the laboratory, as we truly believe that our expected results will improve the effectivity of current immunotherapies and will be translated in the benefit of the patients.

 & Alex Martínez-Sabadell (right)")




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in